
Синдром Ангельмана
Синдром Ангельмана у детей — генетическое заболевание, которое диагностируют еще у маленьких детей, и которое проявляется задержкой физического и интеллектуального развития, дискординацией движений, беспричинным смехом и т. д.
Причины
На вид дети с этой болезнью всегда выглядят счастливыми и позитивными. Синдром был описан впервые в 65-м году 19-го века Гарри Ангельманом, в честь которого и было названо заболевание. Изначально болезнь была названа синдромом счастливой марионетки, но сегодня это название не актуально.
На 10-20 тысяч младенцев рассматриваемую патологию диагностируют один у одного ребенка. В 15 хромосоме теряются копии нормальных материнских генов в процессе деления хромосомы. Иногда синдром возникает, если у отца есть трисомия, дисомия, или если мутировали гены отца. У здоровых детей есть копия 15 хромосомы отца и матери. Если копия генетически изменена, то это становится причиной синдрома Ангельмана.
Возникновение болезни спонтанное, но риски выше, если у кого-то из родителей есть аналогичная болезнь. Если у вас родился ребенок с синдромом Ангельмана, это не говорит о том, что следующее дитя также получит аналогичный диагноз (риск есть, но это всего один процент). Выше риски синдрома Агнельмана, если у родителей есть какие-либо аномалии хромосом в организме.
Симптомы
Основные проявления синдрома:
- плохой набор веса
- плохой аппетит
- задержка или отсутствие речи
- задержки развития
- ребенок понимает мысли нормально, но высказать свои мысли не может
- хаотичные движения руками или ногами (это может быть и тремор, а не полноценное движение)
- значительные трудности в обучении
- больной ребенок не может в достаточной мере концентрировать внимание
- могут быть деформированные черты лица (выдвинутый вперед крупный подбородок, широкий рот, редко расположенные зубы)
- часто дети с данным синдромом страдают эпилепсией
- улыбки и смех без причины
- ребенок ходит на прямых ногах, не сгибая их, как марионетка (потому старое название синдрома именно такое, как уже отмечалось выше)
- уплощение затылка
- проблемы со сном
- маленькая голова
- повышенная чувствительность к жаре
Выше перечисленные признаки вовсе не обязательно присутствуют в полной мере у больного ребенка. Часть из них может быть очень ярко видна, а некоторые могут и отсутствовать. Умственное и физическое развития всегда задерживаются. Проблемы с речью также очень характерны для рассматриваемого синдрома. А судороги и изменение нормальной формы головы встречаются в более редких случаях.

Очень редкие симптомы синдрома Ангельмана:
- высунутый язык, который дрожит
- нижняя челюсть, выдвинутая вперед
- нарушение питания
- слишком активные сухожильные рефлексы
- косоглазие
- бесконтрольно двигающийся язык
- нарушение сосательного и глотательного рефлексов
Проявления синдрома могут меняться по мере взросления ребенка. Во взрослом возрасте больные выглядят моложе своих лет. Половое созревание в определенный момент таки наступает, потому с репродуктивной функцией у них всё в порядке. Такие пациенты могут иметь своих детей. Но есть очень высокий риск, что у их ребенка найдут ту же болезнь.
Во взрослом возрасте при рассматриваемом диагнозе у человека может быть нарушение мелкой моторики и недержание мочи. В основном они носят одежду без застежек, потому что самостоятельно одеться из-за нарушений моторики не могут. Бытовые навыки осваиваются больными в достаточной мере. Они могут сами кушать, без посторонней помощи чистить зубы, стирать вещи и т.д. В большинстве случаев, доживая до зрелого возраста, пациенты получают ожирение и выраженный сколиоз, который с годами усугубляется.
Словарный запас больных детей очень небольшой. Часть из них совсем не разговаривает. Присуща такая черта характера как дружелюбие и коммуникабельность. Дети любят своих родных, проявляют ласку. Желательно ребенка с ранних лет научить общаться через язык жестов. С этой целью созданы специальные программы обучения. Они помогают ребенку совершенствовать моторику, чтобы обслуживать себя в быту и работать.
Лечение
Лекарствами синдром Ангельмана не лечится. Но есть определенные меры, которые помогут детям лучше себя чувствовать и более-менее комфортно существовать в обществе. Если у ребенка обнаружено снижение мышечного тонуса, проводится физиотерапия и массаж для укрепления. При формировании речи ребенка лучше отвести к логопеду. Это поможет в некоторой мере исправить артикуляцию и т.п. При нарушениях сна доктор назначает снотворные, которые облегчат процесс засыпания.
Редко у пациентов бывают судороги. Но, если такое есть, то назначаются противосудорожные лекарства, которые покупаются по рецепту врача. Дети с рассматриваемым синдромом не выглядят плохо. Как уже было отмечено, у них довольный внешний вид, они находят общий язык с окружающими, не являются агрессивными. Но в части случаев коммуникация сложный для них процесс по причине специфического отношения здоровых людей к больным. Иногда установление контакта занимает довольно много времени.
Также нормальной коммуникации ребенка препятствуют недоразвитые навыки речи. Как уже было отмечено, овладев языком жестов, ребенок сможет в достаточной мере выражать свои мысли.
Уход за ребенком и прогноз заболевания
Болезнь обнаруживают в основном, когда ребенок находится в возрасте от трех до семи лет. Симптоматика выражена четко именно к достижению этого возраста, не раньше. Поскольку симптомы неспецифичны, тяжело диагностировать рассматриваемый синдром у младенца. Важно отметить, что болезнь относительно редкая. Потому врач, есть вероятность, не сможет и догадаться, что он имеет дело с синдромом Ангельмана.
В Европе есть больше данных про рассматриваемую патологию, потому диагностика у них более качественная и быстрая. Поставить диагноз должен врач-генетик, который назначает генетический анализ, на основе которого делаются выводы.
Прогноз болезни коррелирует со степенью поражения пятнадцатой хромосомы. Часть пациентов нормально ведут себя в быту, общение у них также почти нормальное. Но в части случаев ребенок не может ходить, речь у него ответствует. Люди с синдромом Ангельмана живут 20-50 лет. Качество жизни во многом зависит от тяжести синдрома, как уже было сказано выше.
Для жизни больного важно понимание и внимание его семьи. Они должны понять ребенка и дать ему всю любовь, какую только могут, чтобы его психическое развития было полноценным. Часть детей приходится отдавать в интернат или специализированную школу. Там они обучаются по специальным программам и учатся жить в обществе.
Синдром Айкарди (Экарди): причины, симптомы, лечение, прогноз

Синдром Айкарди является редким цереброретинальным генетическим расстройством. При данном заболевании мозг, частично или полностью, лишен одной из основных составляющих – мозолистого тела. Это является следствием дефекта Х-хромосомы. В сетчатке происходят изменения, ведущие к инфантильным спазмам.
Мозолистое тело – центральный отдел нервной системы, представленный нервными окончаниями, соединяющими полушария головного мозга.
Сидром Айкарди не является отдельной формой эпилепсии, а является самостоятельным заболеванием, которое в 1965 году было описано невропатологом из Франции Жаном Экарди.
Синдром Айкарди-Гутьереса является угрозой для мальчиков с хромосомным набором ХХY или с синдромом Клайнфельтера.
По данным статистики, на сегодняшний момент наблюдалось около 500 случаев этого заболевания. Наибольшее распространение оно получило в Японии.
Однако, национальности и расы не играют ключевой роли. Все зависит от уровня медицины в регионе. Недостаток знаний ведет к не выявленным случаям. Из 2 – 4% детей, у которых проявляются инфантильные спазмы, обнаруживается болезнь Экарди.
Этиология и патогенез синдрома
До сих пор точно не установлена причина данного заболевания. Соответственно, невозможно определить факторы риска.

Специалисты отвергают наследственность. Если в семье есть ребенок с данным синдромом, то шанс рождения второго с такой же болезнью составляет меньше 1%.
Выдвигается следующая теория. Клетки эмбрионов содержат по одной активной Х-хромосоме. Следовательно, их инактивация при нормальном развитии происходит случайно.
При синдроме Айкарди, случайной инактивации не наблюдается и большинство клеток формируется при участии только этой хромосомы.
Какой ген отвечает за эти изменения к настоящему времени неизвестно. Предположительно, это мутированный de novo на инактивированной Х-хромосоме.
Как проявляет себя синдром?
Дети 2 – 5 месяцев, у которых обнаруживается синдром Айкарди, внешне не отличимы от здоровых малышей. Позже начинают проявляться судороги или инфантильные спазмы. Они представляют вид эпилептического синдрома.
Ребенок снижает активную деятельность и впадает в ступор. Взгляд прикован к одной точке. Руки начинают подниматься вверх и сгибаться.
Тело выгибается и ножки начинают резко выпрямляться. На это уходят секунды и продолжения можно ожидать в любое мгновение. Во время припадков, малыш становится раздражителен и постоянно плачет. Позже, такие судорги часто ведут к эпилепсии.
Параллельно присутствуют и другие симптомы синдрома Айкарди:
На фото глазное яблоко при синдроме Айкарди
По наблюдениям за больными можно выделить и дополнительные признаки:
- латеральное расположение бровей, их редкость;
- выступающие резцы и вздернутый нос;
- уменьшен угол носовой перегородки.
Иногда наблюдаются наросты и уплотнения на коже: невусы, дивертикулы кожи и опухоли, причиной которых является патология кровеносных сосудов – гемангиомы. Редко встречается аномалия рук.
Встречаются заболевшие с плагиоцефалией (сплюснутая область на голове), с ассиметрией лица, а также с расщелинами неба или верхней губы. Размеры головы и рук заметно меньше, чем у здорового человека. Нос плоский, а уши чрезмерно большие. Нос и губы разделяет слишком малое пространство.
У большего количества больных происходило развитие тяжелых эпилептических припадков, которые не проходили до конца жизни. Известно наличие полу-позвонков и отсутствия ребер, приводящее к заметному искривлению позвоночника.
Увеличен риск развития опухолей:
- папиллом сосудистого сплетения;
- липом;
- ангисарком;
- гепатобластом;
- гемангиом;
- ангиосарком;
- гепатобластом;
- интерстициального полипоза;
- эмбриональной кальциомы;
- полипоза кишечника.

Аномалии в глазах могут привести к пигментному рениту, микрофтальми. Возможна катаракта и атрофия зрительного нерва. Это может привести к нарушению зрения и даже полной слепоте.
Нередко нарушается эндокринная система – позднее наступление половой зрелости или явная задержка.
Физическое развитие начинает замедляться. В возрасте 7-10 лет больной выглядит как пятилетний. Вес увеличивается также с задержкой.
Согласно исследованиям ученых, в мозге здорового ребенка больше складок, чем у болеющего синдромом Айкарди. Случается, в пораженном мозге образуются кисты, которые заполнены жидкостью.
Большинство заболевших имеют явно выраженную задержку в умственном развитии, однако, иногда они просто плохо способны к обучению.
Способность к членораздельной речи часто развита очень слабо. Самостоятельное передвижение наблюдается редко. Некоторые полностью зависимы от помощи других людей.
Постановка диагноза
На сегодняшний момент нет специальной технологии диагностики, с помощью которой можно точно поставить диагноз. Как правило, применяются следующие методы:
- Неврологический осмотр, в процессе которого врач оценивает состояние нервной системы ребенка и определяет ее расстройства.
- Офтальмоскопия включает в себя изучение глазного дна, его сосудов и сетчатки.
- Электроэнцефалограмма (ЭЭГ). С ее помощью исследуют работу головного мозга, происходит регистрация электрических импульсов, которые исходят из его отдельных частей. Является основным методом для диагностики эпилепсии и других заболеваний. При синдроме Айкарди помогает определить тяжесть и тип приступов.
- Магнитно-резонансная томография (МРТ). Основу этого диагностического исследования составляет постоянное магнитное поле, быстро меняющиеся локальные магнитные поля и радиочастотная энергия. Специально предназначенная для этого аппаратура создает четкое изображение внутреннего органа. С помощью обнаруживается патология мозолистого тела, ассиметрия полушарий, внутримозговая киста и другие аномалии.
- Компьютерная томография (КТ). С ее помощью специалисты выявляют участки мозга, которые подверглись повреждениям в результате болезни.
- Рентген скелета.
- Генные исследования.

Другие диагностические методы могут проводиться в зависимости от симптомов и общего состояния больного.
Симптоматическое лечение — единственный вариант

На сегодняшний момент комплексного лечения синдрома Айкарди нет. Применяется терапия каждого симптома.
Принятая тактика – купировать инфантильные спазмы, являющиеся резидентными к антиэпилептическим препаратам. Используются максимально высокие дозы разнообразных медикаментов.
После выявления заболевания, на первых порах используется Вигабатрин, он же Сабрил. Его применяют от 50 до 100 мг на килограмм веса в сутки. Параллельно с ним вводятся вальпроаты. Их суточная норма – 50–100 мг на кг.
Если приступы учащаются, то к АЭП добавляются бензодиазепины, от 0,25 до 2 мг в сутки. Нередко применяется Фенобарбитал или Суксилеп.
В качестве альтернативы выступают кортикостероидные гормоны. Это:
- АКТГГ;
- Синактен-депо;
- Дексаметаз;
- Преднизолон;
- Октагам.
Поначалу Преднизалон назначается из рассчета 1–2,5 мг на кг, затем эта доза снижается до щадящей. Как правило, гормоны применяются параллельно базовым АЭП.
К сожалению, эти методы малоэффективны и редко достигают полного подавления синдрома.
В виде палиативного хирургического лечения может быть стимулирован блуждающий нерв. Дефекты костей, которые способствуют сколиозу, устраняются физиотерапией, лечебной физкультурой или исправляются при помощи хирургической коррекции.
Разработаны специальные программы, противодействующие умственной отсталости. Для длительного лечения патологических состояний необходима помощь детского невропатолога.

Прогноз и летальность
Прогноз при синдроме Айкарди достаточно неблагоприятный и зависит от тяжести спазмов и сопровождающих их заболеваний:
- смертность в детском возрасте – 25%;
- выживают и могут самостоятельно ходить – 25%;
- могут самостоятельно себя обслуживать – 50%.
Больные живут приблизительно от 9 до 19 лет. Однако известны случаи, когда заболевшие женщины прожили до 32 и 49 лет (умеренная форма синдрома).
На нем можно пообщаться с другими людьми, страдающими этим заболеванием, и их семьями. Задать интересующие вопросы, поделиться опытом и другое. Здесь можно получить информацию о благотворительных мероприятиях, организатором которых является Фонд Экарди.
К сожалению, в связи с мало изученностью данного заболевания, профилактических мер против синдрома Айкарди пока не существует.
Синдром Ангельмана: патогенез, тип наследования, симптомы, фото детей, лечение
Синдром Ангельмана (ранее известный как «синдром счастливой марионетки») включает судорожные движения, беспричинный смех и задержку умственного развития различной, чаще всего серьезной умственной отсталости (Angelman 1965; Horsier и Oliver 2006). Частота в популяции составляет около 1 на 12000 живых новорожденных, с соотношением мужчин и женщин 1:1 (Steffenburg et al., 1996). Исчерпывающее описание синдрома Ангельмана в настоящее время находится в печати (Dan, 2008).
а) Патогенез. Синдром Ангельмана в большинстве случаев вызван делецией 15q11.2-12 хромосомы, сходной, но не идентичной выявляемой у детей с синдромом Прадера-Вилли (Magenis et al., 1987, Knoll et al., 1989) и наследуется по материнской линии (Knoll et al., 1989). Делеция включает ген бета-3-субъединицы ГАМК-рецептора (Saitoh et al., 1994). Среди 60-75% пациентов отмечаются делении или перестройки длинного плеча 15-й хромосомы, делеция всегда располагается на материнской хромосоме.
В небольшом количестве случаев выявляется дисомия отцовской 15-й хромосомы (Malcolm et al., 1991, Prasad и Wagstaff, 1997). Однако не менее чем у 15% пациентов выявляются нормальные хромосомы и отсутствуют признаки дисомии. В некоторых случаях такого рода возможно повторное рождение больных детей среди родственников (Clayton-Smith, 1992). Такие случаи могут быть связаны с доминантной мутацией гена UBE3A (Kishino et al., 1997) на 15q11-13 хромосоме, приводящей к возникновению фенотипа Ангельмана только при передаче по женской линии (Wagstaff et al., 1993).
Различия клинических проявлений зависят от происхождения генетического дефекта (Lossie et al., 2001). Вызванные делециями формы, обычно имеют более выраженный характер, чем формы, вызванные единичной мутацией гена или другими генетическими дефектами. Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
б) Диагностика. Диагноз подтверждается в случае, если клинический фенотип соответствует положительному FISH-тесту. У младенцев (van Lierde et al., 1990, Yamada и Volpe, 1990) даже при использовании предложенных критериев диагноз зачастую затруднен (Williams et al., 1995). Весьма вероятным диагноз представляется при сочетании задержки умственного развития с проявлениями аутизма, жизнерадостным поведением, атаксией и эпилептическими приступами (часто «минимально» выраженными).
На основании апраксической походки, стереотипиях движений рук и (в некоторых случаях) гипервентиляции возможна ошибочная постановка диагноза синдрома Ретта у девочек. Тем не менее, при раннем (до года) начале припадков, жизнерадостном настроении и дисморфизме вероятность неверной диагностики можно исключить.
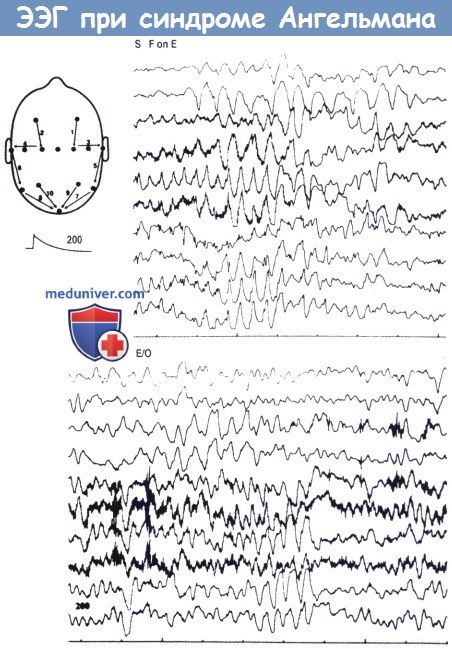
Типичные изменения на ЭЭГ двух детей различного возраста (верхний рисунок —семь лет, нижний рисунок — два года), страдающих синдромом Ангельмана.
Следует обратить внимание на калибровку. В обоих случаях отмечается повышенная ритмичная тета-активность, чередующаяся с эпизодами медленного ритма, в частности, в области передней половины двух полушарий.
На верхнем рисунке эпизоды активности с частотой 3-4/с, чередующиеся со слабовыраженными острыми волнами, отмечаются в задней трети головы в случае, когда помощник закрывает пациенту глаза.
S — закрыты, F on Е — «пальцы закрывают глаза», Е/О — глаза открыты.
в) Клинические проявления. Клинические проявления включают серьезную умственную отсталость с тяжелым нарушением речи, атаксию и заметные спастические нарушения движений верхних конечностей, а иногда и туловища; данные двигательные нарушения являются особым типом миоклонуса (Guerrini et al., 1996; Beckung et al., 2004). Немотивированные приступы смеха являются характерным, но не основным проявлением, в то время как жизнерадостное настроение является постоянным признаком (Williams и Frias, 1982).
У 86% больных детей отмечаются приступы, которые часто имеют повторный характер, но длятся недолго и чаще всего являются не тонико-клоническими припадками, а атипичными абсансами или тоническими или атоническими припадками (Dorries et al., 1988; Viani et al., 1995; Laan et al., 1997). На ЭЭГ часто встречаются характерные изменения в виде эпизодов медленных (3 Гц) волн, особенно в задних отделах, часто зазубренных и в отдельных случаях связанных с истинными пиками (Boyd et al, 1988).
На КТ и МРТ каких-либо специфичных изменений не выявляется, но возможно наличие небольшого расширения желудочков и/или околомозгового пространства (Dorries et al., 1988).
Способность к передвижению появляется поздно, часто после 5-6 лет; походка имеет аномальный характер с широко расставленными ногами и атаксическими и апраксическими признаками (Sugimoto et al., 1992).
Дисморфический синдром характеризуется умеренной выраженностью и может быть незаметен в детском возрасте. Прогнатизм обычно развивается только в возрасте нескольких лет.
Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
Только недавно были опубликованы результаты исследований, подтверждающих мнение о специфическом поведенческом фенотипе данного синдрома, и еще слишком рано делать предположения в данном направлении. Результаты одного из исследований взрослых и молодых пациентов, страдающих синдромом Ангельмана (Clayton-Smith, 1993), свидетельствуют о том, что гиперактивность, часто наиболее выраженная в раннем детском возрасте, в позднем детском возрасте обычно сменяется более контролируемым поведением.
Результаты большинства исследований в настоящее время позволяют предположить, что мутизм может быть постоянным проявлением, но некоторые пациенты могут освоить язык жестов. В одном из центров наблюдался мальчик с сочетанием синдрома Ангельмана и аутизма. У многих детей с синдромом Ангельмана отмечаются заметные аутистические проявления, такие как упрямство, настаивание на одном и том же и увлеченность водой, и в то же время они кажутся счастливыми и общительными, так как им нравится контактировать с другими людьми. Тем не менее, контакт осуществим лишь при выполнении их собственных условий.
г) Исход. Прогноз неблагоприятный, отмечается серьезная умственная отсталость. Овладение коммуникативным языком невозможно (Beckung et al., 2004).
Синдром Ангельмана

Синдром Ангельмана (СА) – генетическое заболевание, которое вызвано хромосомными нарушениями. Оно имеет несколько названий, отражающих проявления болезни: синдром счастливой куклы, а также марионетки или Петрушки
Синдром Ангельмана у детей становится заметным в первые годы жизни. При рождении ребенок с генетическим дефектом кажется абсолютно здоровым. Первые симптомы проявляются к годовалому возрасту.
При подобной патологии наблюдаются неврологические расстройства, отставание в психическом развитии, возникают двигательные трудности, проявляются симптомы эпилепсии.
Синдром счастливого Петрушки характеризуется следующими признаками: радостное лицо, приступы беспричинного смеха, марионеточная походка. Такие дети доброжелательны, эмоциональны, любят общение, часто смеются, всегда улыбаются.

Синдром Ангельмана: клинические симптомы
Впервые болезнь описал английский педиатр Г. Ангельман. Ее признаки бывает трудно определить до года. Явно выраженные симптомы проявляются с двухлетнего возраста. К этому времени уже заметны клинические проявления:
1. задержка психического развития – снижение когнитивных функций, умственная отсталость
2. речевые нарушения – ограниченная речь или ее отсутствие
3. психомоторные расстройства – хаотичные движения рук, тремор конечностей, атаксия (проблемы с координацией), специфическая походка
4. нарушения физического развития – черепно-лицевые аномалии: микроцефалия (объем черепа меньше нормы), редкие зубы, слюнотечение, уплощенный затылок, выступающая нижняя челюсть, открытый рот, высунутый язык. часто наблюдается сколиоз, мышечная дистония, косоглазие
5. эпилептическая активность, приступы судорог
6. особенности поведения (гиперактивность, смех без повода)
7. беспокойный сон.
Синдром Ангельмана у взрослых может быть менее выражен. С возрастом клинические проявления ослабевают – снижается гиперактивность, улучшается сон, уменьшается частота судорожных приступов. У них часто проявляются ожирение и сколиоз.
Внешние признаки при легкой форме болезни могут быть незаметными. Многие взрослые выглядят значительно моложе своих лет. Из-за выраженной задержки в развитии они в душе остаются детьми – могут быть импульсивными, чрезмерно обидчивыми, а иногда агрессивными.
Синдром Ангельмана: основные причины заболевания
Различают несколько вариантов генетической патологии при СА. Чаще всего причина заболевания – генная мутация определенного участка 15 хромосомы матери. Аномалия обусловлена делецией – потерей хромосомного сегмента.
Могут быть и другие причины:
• трисомия – наличие лишней хромосомы
• микроделеция – отсутствие одного гена
• инверсия – нарушение генного порядка из-за разворота отдельного участка хромосомы
• транслокация – перенос части одной хромосомы к другой.
 Диагностика
Диагностика
 Диагностика
ДиагностикаМногие пациенты наблюдаются у специалистов с эпилепсией, речевой задержкой, различными нарушениями поведения, не зная о своем заболевании. Для постановки диагноза необходима консультация невролога, психиатра, врача-генетика.
Выявлять аномалии, которые приводят к возникновению синдрома Ангельмана, позволяют исследования: генетический анализ, МРТ, ЭЭГ, УЗИ. Самыми надежными считаются генетические исследования. На основании их результатов при характерном проявлении симптомов заболевания можно установить точный диагноз.
Диагностировать АС можно еще до рождения ребенка во время беременности. Для этого применяют два вида исследований:
1. неинвазивные – скрининги, включающие комплексное исследование по выявлению у плода хромосомных аномалий. Достоверность результатов не всегда подтверждается
2. инвазивные – высокоточные методы, их назначают женщинам старше 35 лет для подтверждения результатов, полученных другими методами. Они небезопасные, проводятся только по показаниям, учитывая высокий риск осложнений.
Лечение и профилактика
Влиять на генетический дефект невозможно, нарушения происходят на хромосомном уровне, поэтому больным назначают симптоматическое лечение и психологическую коррекцию.
Медикаментозная терапия может применяться при эпилептических приступах. Для смягчения симптомов заболевания и улучшения сна могут использоваться успокаивающие препараты.
Разрабатываются индивидуальные программы для каждого пациента – ЛФК, логопедические занятия, лечебный массаж, поведенческая терапия. Они дают возможность улучшить качество жизни человека. Также привлекают логопедов, дефектологов, специалистов по невербальным способам общения и поведенческой терапии.
В профилактических целях парам, планирующим зачатие ребенка, рекомендуется консультация генетика.
Прогноз
Тяжесть болезни может быть разной. Прогноз зависит от степени повреждения материнской 15 хромосомы. Некоторые пациенты могут себя обслуживать и общаться, другие – не способны передвигаться и говорить.
Проведение физиотерапии для развития точности движений, коррекция коммуникационных навыков улучшают прогноз. Но в любом случае больные не смогут стать абсолютно самостоятельными, им требуется помощь на протяжении всей жизни.
Синдром Ангельмана ( Синдром марионетки , Синдром Петрушки , Синдром счастливой куклы )
Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития. Проявляется интеллектуальным отставанием, слабой сформированностью речи, навыков сидения и ходьбы, хаотичными движениями, гиперактивностью, симптоматической эпилепсией, беспричинным весельем и смехом, сколиозом, своеобразной походкой. Пациенты имеют особую внешность: рот крупный, зубы расположены редко, подбородок выдается вперед. Диагноз устанавливается на основании клинических данных, результатов генетического анализа. Специфическое лечение отсутствует, проводится симптоматическая терапия, оказывается психологическая и педагогическая помощь.
МКБ-10
Общие сведения
Синдром назван по фамилии британского педиатра Г. Ангельмана. В 1965 году он первым описал симптомы заболевания и назвал его «синдромом счастливой марионетки», поскольку пациенты напоминали ему героя картины «Мальчик-марионетка». В те годы методы генетических исследований были еще не разработаны, установить причину патологии было невозможно. В 1987 году исследователи определили этиологию болезни и переименовали ее в синдром Ангельмана. Сейчас этот термин является официальным, но можно встретить синонимичные названия – «синдром марионетки», «синдром Петрушки», «синдром счастливой куклы». Распространенность составляет 1 случай на 10-20 тысяч новорожденных. Заболевание выявляется после первого года жизни (иногда – к 3-7 годам), чаще болеют мальчики.
Причины
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но его характер и способ возникновения могут различаться. Иногда заболевание дебютирует в результате передачи измененной генетической информации от родителя, иногда является последствием спонтанных нарушений в геноме. Хромосомные аномалии удается определить примерно у 85-88% больных. Причиной синдрома может быть:
- Делеция. При данном дефекте часть генетического материала теряется или инактивируется. У 70% пациентов диагностируются обширные делеции области 15q12 хромосомы, в которой локализован активатор гена.
- Однородительская дисомия. ОРД определяется в 2-3% случаев болезни. В хромосомном наборе присутствуют две копии 15 хромосомы отца. Материнской хромосомы нет, ген также отсутствует.
- Дефект запечатления. Суть аномалии заключается в том, что центр запечатления, регулирующий активность локуса UBЕ3A, оказывается нефункциональным, «выключенным». Ген остается структурно целым, но не выполняет своих функций. Распространенность ДЗ – 3-5%.
- МутацияUBE3A. У 5-10% пациентов причиной болезни являются мутационные изменения гена. Они представлены инверсиями, микроделециями, транслокациями и дупликациями.
Патогенез
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген кодирует производство протеина Е6АР, который представляет собой ферментный компонент сложной реакции деградации белков. Е6АР участвует в процессе образования убиквитина – белка системы протеасом, стимулирующего протеолиз дефектных белковых молекул в нейронах головного мозга. В норме убиквитин маркирует ненужные (неактивные, нефункциональные) белки с целью инициации их уничтожения. Е6АР обеспечивает закрепление убиквитина на поверхности молекулы белка-мишени. Потом протеасомы расщепляют его на пептидные остатки и на аминокислоты. При синдроме Ангельмана убиквитин не закрепляется на дефектных белках, они скапливаются в нервной ткани мозга, нарушается процесс синаптической передачи. Формируются отклонения, задержки в психическом и моторном развитии.
Симптомы
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее освоенные навыки сохраняются, но приобретение новых происходит медленно. Дети умеют сидеть, ползать, брать предметы и перекладывать их из руки в руку, поддерживать визуальный контакт, гулить и лепетать. Ходьба дается с трудом, нарушено чувство равновесия, наблюдаются частые падения, ушибы о мебель. Выявляется тремор и хаотичные движения конечностями, особенно руками. Речевые расстройства представлены как задержками, так и полным отсутствием экспрессивной речи. Дети либо совсем не говорят, либо используют лепет, простые слоги и слова общим объемом не более 10 единиц. Сохраняется понимание обращенной речи, стремление к общению, использование невербальных средств коммуникации: жестов, мимики, опосредованных знаков.
Основное поведенческое нарушение – гиперактивность. Дети часто веселятся и смеются без объективной причины, двигательно расторможены, неусидчивы, нецеленаправленны. У многих возникает патологическая привязанность к определенной игрушке или предмету быта, при появлении которого настроение сразу повышается, капризность и плач сменяются смехом. Концентрация внимания снижена, переключаемость быстрая и ненаправленная. Имеются трудности обучения, стойкое снижение интеллектуальных функций. Легко закрепляются стереотипии: раскачивание тела, размахивание руками. У 80% пациентов отмечается микроцефалия, недостаточный объем черепной коробки, эпилептическая активность мозга. Редко наблюдается снижение контроля движений языка, которое проявляется трудностями сосания груди или соски, последующим недостатком массы тела.
Характерные особенности внешности детей – косоглазие, сколиотическое искривление позвоночника, увеличение зубов и губ, разряжение зубного ряда, уплощение затылка, выступание вперед подбородка. Язык часто высунут, рот приоткрыт в улыбке. Развивается мышечная дистония, выраженность рефлексов сухожилий повышается, формируя специфичность моторики: пациенты ходят на прямых несгибающихся ногах, плечи приподнимают, руки сгибают в локтях. Своеобразный симптом – тяга к воде. Большинство детей чувствуют себя спокойнее в водной среде, им нравится плескаться в ванной, играть с корабликами в тазу.
Осложнения
Синдром Ангельмана – редкое малоизвестное заболевание. В связи с этим постановка диагноза и оказание медико-психолого-педагогической помощи зачастую проводятся несвоевременно, к 6-8 годам, что обуславливает низкую успешность коррекционных и лечебных мероприятий. Без физиотерапевтического лечения усугубляются нарушения опорно-двигательного аппарата – больные страдают от тяжелых форм сколиоза, самостоятельно передвигаются с трудом. Своеобразие внешности и поведения становится причиной ухудшения и без того затрудненной социальной адаптации. Все перечисленное влечет за собой утяжеление инвалидизации пациентов.
Диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы на отсутствие речи, двигательные стереотипии, трудности формирования ходьбы и других двигательных навыков, гиперактивность. Проводится дифференциальная диагностика, в ходе которой должны быть исключены более распространенные заболевания, такие как умственная отсталость, расстройства аутистического спектра, деменции, мутизм, несимптоматические формы эпилепсии. Комплексное исследование включает следующие процедуры:
- Общий осмотр. На наличие синдрома часто указывают специфические черты внешности пациентов: высунутый язык, слюнотечение, крупный рот, широкие редкие зубы, выступающая вперед нижняя челюсть, плоская форма затылка. Характерен светлый оттенок кожи, глаз и волос. Походка детей напоминает движения куклы-марионетки из-за усиления сухожильных рефлексов и снижения мышечного тонуса.
- Осмотр психиатром. В 100% случаев синдрома Ангельмана диагностируется выраженная задержка в развитии психики, отсутствие самостоятельной речи или очень скудный словарный запас. Коммуникация осуществляется с помощью мимики, жестов, рисунков. В поведении отмечается гиперактивность, стереотипные движения руками, беспричинный смех.
- Неврологическое обследование. У всех пациентов определяется атаксия и тремор конечностей. 80% больных имеют постнатальную микроцефалию – окружность головы новорожденного меньше 32 см, к 12 месяцам – около 42 см. По данным ЭЭГ выявляется симптоматическая эпилепсия (высокоамплитудные разряды медленных комплексных волн), клинически возможны судорожные припадки. У некоторых детей имеется косоглазие, диффузное снижение мышечного тонуса, усиление сухожильных рефлексов, гиперкинезы.
- Генетическое исследование. Лабораторная диагностика нацелена на выявление мутаций и делеций в гене UBE3A. Последовательно проводится комплекс процедур, включающий флуоресцентную гибридизацию in situ, анализ мутации центра запечатления, метилирование ДНК СА/ПВС региона, диагностику делеции методом микроматричного анализа, поиск мутационных изменений в локусе UBE3A.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
Прогноз и профилактика
Выраженность симптомов синдрома Ангельмана может сильно различаться. Пациенты с легкими формами болезни имеют благоприятный прогноз: их речь становится более развернутой, улучшаются навыки самоконтроля при некоторых нарушениях двигательной сферы. При любой степени тяжести раннее начало и регулярное проведение медико-психологической помощи повышает качество жизни больных. Профилактика сводится к генетическому обследованию пар, в семьях которых есть ребенок с данным синдромом. Характер хромосомного дефекта (спорадический или наследственный) позволяет определить риск рождения второго больного ребенка.
2. Генетические механизмы возникновения синдрома Ангельмана и их фенотипические проявления / Отрошко Е.В., Молодожникова Н.В. // XXIV Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки» – 2015.
Синдром Ангельмана
Синдром Ангельмана – это генетическое заболевание, сопровождающееся отставанием в психическом развитии. Болезнь сочетается с припадками, с немотивированным смехом и хаотическими движениями рук. Подобные проявления у больных людей способствовали тому, что синдром Ангельмана часто называют синдромом Петрушки или синдромом смеющейся куклы, либо счастливой куклы.
Эпоним данный синдром получил от врача-педиатра Б. Г. Ангельмана, который впервые описал ее в 1965 году. Согласно статистике, синдром Ангельмана встречается у 1 ребенка из 10-20 тысяч.
Симптомы синдрома Ангельмана
Симптомы синдрома Ангельмана многообразны.
У некоторых больных они проявляются чаще, у других реже, но в целом клиническая картина выявляется следующая:
Внешние признаки: окружность головы больного меньше средних значений, зубы расположены редко, затылок уплощен. Разрез рта у больного широкий, из него виден язык, который часто высунут наружу, подбородок при этом выдается вперед. Иногда диагностируется косоглазие.
Сразу после рождения начинаются проблемы с питанием. Дети плохо кушают, вес набирают с трудом.
Всегда присутствует ЗПР, в раннем возрасте дети поздно начинают садиться и ходить.
Ребенок хорошо понимает, что до него пытаются донести, но высказать свою мысль может с трудом. Страдает речевое развитие.
Гиперактивность – характерная черта таких детей. Движения конечностями при этом хаотичны, заметен мелкий тремор.
Страдает внимание, в первую очередь такой процесс, как концентрация. Это приводит к проблемам в обучении.
Нередки эпилептические припадки.
Улыбка ребенка беспричинная, смех немотивирован.
Характерна для больных с синдромом Ангельмана марионеточная походка, то есть ноги во время движения сгибаются в коленных суставах минимально.
Всегда имеются проблемы со сном.
Больные плохо переносят высокие температуры, хорошо чувствуют себя в воде.
Если рассматривать частоту встречаемости симптомов, то у больных всегда имеются проблемы с психическим и физическим развитием, страдает речь, поведение, моторика, концентрация внимания. В 80% случаев у детей случаются приступы эпилепсии.
Реже наблюдаются судороги, деформация черепа. Совсем нечасто выявляются проблемы с питанием, косоглазие, дрожащий язык.
Случается, что по мере взросления больного, симптоматика несколько меняется. При этом все люди с синдромом Ангельмана выглядят младшего своего возраста. Некоторые из обзаводятся семьями, однако риск рождения ребенка с аналогичной патологии высока.
С возрастом у больных прогрессирует сколиоз, имеется склонность к ожирению.
Причины синдрома Ангельмана
Причины синдрома Ангельмана кроются в генетических аномалиях, так как в 15 хромосоме у больных людей отсутствует часть генов. Эти хромосомные нарушения объясняются явлением делеции, либо мутациями.
Синдром Ангельмана развивается в том случае, когда нарушения происходят в материнской хромосоме, если изменяется отцовская хромосома, то у больного диагностируют синдром Прадера-Вилли.
Ведущей причиной патологии называют нарушения в делении 15 хромосомы, которые влекут за собой аномалии в создании копий материнских генов. Реже к развитию болезни приводит мутация отцовских генов, отцовская трисомия и дисомия.
Так, здоровые люди получают от обоих родителей по 1 копии 15 хромосомы, когда ребенок получает от одного из родителей мутированную копию (чаще всего от матери), то у него развивается данный синдром.
Диагностика синдрома Ангельмана
Диагностика синдрома Ангельмана выстраивается на основе проведения генетического исследования 15 хромосомы. Этот анализ показан детям с гипотонусом, с нарушениями психического и физического развития.
Основанием для генетического анализа служат симптомы, характерные для синдрома Ангельмана. Стоит отметить, что существует отдельная группа людей, у которых все анализы остаются в норме, а признаки болезни присутствуют.
Лечение синдрома Ангельмана

Лечение синдрома Ангельмана сводится к выполнению определенных мероприятий, которые позволяют улучшить качество жизни больного. Медикаментозная терапия является бессильной, так как нарушения происходят на хромосомном уровне. Поэтому в детстве показан лечебный массаж для коррекции гипотонуса, а также выполнение укрепляющих физиопроцедур.
По мере взросления ребенка, ему необходима помощь логопеда. Регулярные занятия позволяют добиться более четкого произношения и артикуляции.
При проблемах со сном, рекомендуют к приему снотворные препараты. Так, пациентам до ночного отдыха (за 30 минут), рекомендуют прием мелатонина в дозировке 0,3 г. Эпилептические припадки, тремор конечностей и судороги с успехом купируются противосудорожными средствами.
Легкие слабительные препараты назначают при проблемах со стулом.
Врачи в США проводят терапию синдрома Ангельмана с помощью гормона Секретин, который используется для лечения детей с аутизмом. Препарат вводится внутривенно и способствует снижению симптомов заболевания, нормализуя поведение и улучшая коммуникативные навыки.
При этом, люди с синдромом Ангельмана не ущербны: они очень общительны и дружелюбны, хорошо ладят с детьми, всегда в хорошем настроении. Недостаточность коммуникативных навыков возможно заместить невербальными средствами общения.
Что касается прогноза, то высок риск того, что второй ребенок в семье будет также страдать синдрома Ангельмана, поэтому, чтобы этот риск минимизировать, необходима консультация у генетика. Дело в том, что если мутация не унаследованная, то вероятность появления второго больного ребенка сводится всего к 1%.
Прогноз на жизнь в целом благоприятный, хотя зависит от степени поражения хромосомы. Так, некоторые больные полноценно осваивают навыки самообслуживания, способны общаться с окружающими людьми. Иные пациенты, напротив, не способны контролировать даже собственный стул, не в состоянии самостоятельно передвигаться и разговаривать.
Во взрослом возрасте проблему представляет сколиоз, а также ожирение со всеми последствиями этого состояния.

Автор статьи: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность – “Лечебное дело” в 1991 году, в 1993 году “Профессиональные болезни”, в 1996 году “Терапия”.
Наши авторы

