
Синдром Альпорта: причины, симптомы, классификация, лечение, прогнозы
Синдром Альпорта — генетически гетерогенное заболевание, в основе которого лежат мутации генов коллагена IV типа — основного компонента базальной мембраны. Соответственно, клинические проявления синдрома Альпорта связаны с нарушениями в строении и функционировании базальной мембраны. Основной симптом — стойкая микрогематурия.
У мальчиков и мужчин неизбежно развиваются протеинурия, артериальная гипертония и почечная недостаточность, тогда как у девочек и женщин почечная недостаточность возникает редко. Поражение почек нередко сочетается с нейросенсорной глухотой и поражением глаз, в частности дегенерацией пигментного эпителия в области желтого пятна сетчатки и лентиконусом.
Примерно в 80% случаев синдром Альпорта наследуется Х-сцепленно и вызван мутацией гена COL4A5, кодирующего а5-цепь коллагена IV типа. Вариант Х-сцепленного синдрома Альпорта с диффузным лейомиоматозом вызван делецией, затрагивающей лежащие рядом гены COL4A5 и COL4A6. У оставшейся части больных заболевание наследуется аутосомно-рецессивно и связано с мутациями генов COL4A3 или COL4A4, расположенных на 2-й хромосоме и кодирующих а3- и а4-цепи коллагена IV типа соответственно.
Аутосомно-доминантный тип синдрома Альпорта встречается редко, он также связан с мутациями генов COL4A3 и COL4A4.
Существует шесть изомеров а-цепи коллагена IV типа, их обозначают от а 1 до а6. Цепи а.1 и а2 присутствуют во всех базальных мембранах, тогда как экспрессия остальных цепей в разных тканях различна. Цепи с аЗ по а5 имеются в тех органах, которые поражаются при синдроме Альпорта: почках, глазах и ушах. В почках а3-, а4- и а5-цепи присутствуют в базальных мембранах клубочков, дистальных канальцев и собирательных трубочек, а также в капсуле клубочка.
У большинства мальчиков с Х-сцепленным синдромом Альпорта в почечных базальных мембранах полностью отсутствуют цепи с а3 по а6. У девочек с этим заболеванием из-за случайного характера инактивации Х-хромосомы экспрессия этих цепей частично сохранена. При аутосомно-рецессивном синдроме Альпорта аЗ- и а4-цепи отсутствуют во всех базальных мембранах, а5-цепь отсутствует только в базальной мембране клубочка, а в капсуле клубочка и базальных мембранах дистальных канальцев, собирательных трубочек и эпидермиса экспрессия а5- и а6-цепей сохранена. В глазах цепи с а3 по а5 имеются в базальных мембранах капсулы хрусталика, роговицы, задней пограничной пластинки, базальной пластинки (мембраны Бруха) и внутренней пограничной мембраны сетчатки. Они присутствуют также во внутреннем ухе: основной мембране внутреннего уха и базальных мембранах спирального выступа, спирального гребешка, внутренней и наружной спиральных борозд.
Сегодня основной метод диагностики синдрома Альпорта — биопсия почки, хотя, возможно, в будущем ее заменят молекулярно-генетические методы. При электронной микроскопии выявляют расслаивание и утолщение базальной мембраны клубочка. Дополнительное исследование аЗ-, а4- и а5-цепей в ткани почки может оказаться полезным в тех случаях, когда данные биопсии сомнительны. Отсутствие а5-цепи в базальной мембране эпидермиса указывает на Х-сцепленный синдром Альпорта, в этом случае биопсия почки необязательна.
Лечения не существует. Х-сцепленный синдром Альпорта встречается у собак, он служит моделью для разработки генотерапии и лекарственных средств. Трансплантация почки, как правило, дает хороший результат. Из-за того что вместе со здоровой почкой больные получают и новые антигены, примерно у 5% больных развивается антительный гломерулонефрит пересаженной почки, который почти всегда приводит к гибели трансплантата.
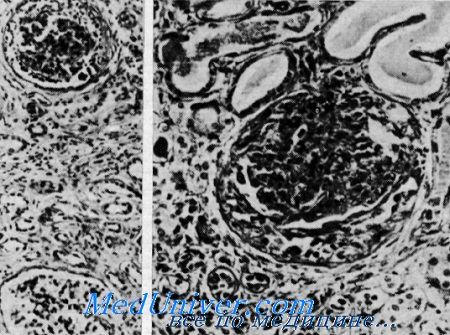
Нефрит и нейросенсорная глухота (синдром Альпорта). Сверху клубочек с утолщенной основной мембраной и слипшейся капсулой в противоположность почти нормальному клубочку внизу. Видны редкие клубочки, имеющие полулупиую форму, расширенные канальцы, содержащие белковые цилиндры. Утолщенная основная мембрана, окружающая атрофичные канальцы в области фиброза (из H. I. Krickstein et al.).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Альпорта: можно ли вылечить или избежать?
Синдром Альпорта являет собой наследственное заболевание, которое проявляется ранним развитием почечной недостаточности, снижением остроты слуха и зрения.
Болезнь обусловлена генетическими мутациями, затрагивающими соединительную ткань – коллаген 4 типа, который является составляющей частью многих важных структур организма, в том числе почек, внутреннего уха и глаз.
Синдром Альпорта намного тяжелее переносится представителями мужского пола. Дело в том, что заболевание чаще всего передается через мутированную Х-хромосому. Так как у девочек две Х-хромосомы, то здоровая срабатывает в качестве запасной и облегчает течение болезни.
При синдроме Альпорта из-за неспособности почек устранять токсины возникает отравление организма. Поэтому у представительниц женского пола эта патология может вызывать бесплодие. А если беременность и наступает, токсины могут погубить и ребенка, и мать. Часто синдром Альпорта проявляется именно во время беременности, если даже ранее не давал о себе знать.
Симптоматика заболевания
Как говорит о таком недуге как синдром Альпорта Википедия, эта наследственная болезнь характеризируется гематурией (кровью в моче), лейкоцитурией (выявление лейкоцитов в анализе мочи), протеинурией (наличие белка в моче), глухотой или тугоухостью, иногда катарактой и развитием почечной недостаточности в подростковом возрасте. Иногда поражение почек может проявляться только после 40-50 лет.
Основной симптом недуга – это наличие крови в моче, что указывает на заболевание почек. Иногда ее можно выявить лишь микроскопически, а в некоторых случаях моча может становиться розовой, коричневой или красной, особенно на фоне подсоединившихся инфекций, гриппа или вирусов в организме. С возрастом кроме гематурии в моче появляется белок и у больного наблюдается артериальная гипертензия.
Хотя синдром Альпорта Википедия описывает как болезнь, которая проявляется катарактой, это не всегда так. Иногда также может возникать аномальная пигментация сетчатки, что значительно ухудшает зрение. Кроме этого, роговица при таком наследственном недуге склонна к развитию эрозий. Потому им нужно беречь глаза от попадания у них инородных предметов.
Синдром Альпорта характеризирует также потеря слуха, которая обычно проявляется в подростковом периоде. Эта проблема решается с помощью слухового аппарата.
Синдром Альпорта: лечение и профилактика
Синдром Альпорта, лечение которого в основном симптоматическое, предполагает обязательную санацию хронических очагов инфекций. Пациентам с этим заболеванием противопоказано проводить прививки в спокойное от эпидемий время. Также имеются противопоказания к приему глюкокортикоидных лекарственных препаратов. При почечной недостаточности применяют диализ, а ее развитие после 20-летнего возраста – это показание для трансплантации почек.
Относительно профилактики патологии, то следует беречься от инфекций мочеполовых путей, которые ускоряют развитие почечной недостаточности. Женщинам с синдромом Альпорта, решившим родить ребенка, необходимо предварительно проконсультироваться с генетиком, который поможет выявить носителя мутантного гена. Хотя статистика показывает, что у около 20% семей, столкнувшихся с синдромом Альпорта, нет родственников, страдающих почечной недостаточностью. Этот факт доказывает, что мутированный ген может возникать спонтанно.
Чтобы уберечь своих потомков от такого наследственного недуга как синдром Альпорта, необходимо избегать родственных браков. А если носитель аномального гена выявлен, чтобы искоренить патологию в будущем, можно воспользоваться донорским генетическим материалом и прибегнуть к процедуре инсеминации или искусственного оплодотворения. В каждом отдельном случае необходима индивидуальная консультация специалистов.
Синдром Альпорта ( Гематурический нефрит , Наследственный нефрит 1 типа , Семейный гломерулонефрит )
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
МКБ-10
Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН. У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам. Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
1. Наследственный нефрит (синдром Альпорта) / Сунгатуллина И.Л. // Казанский медицинский журнал – 2002 – Т.83, №1.
2. Проект клинических рекомендаций по диагностике и лечению синдрома Альпорта у детей / Длин В.В., Игнатова М.С., Конькова Н.Е. – 2014.
3. Наследственные заболевания почек, протекающие с гематурией / Длин В.В., Игнатова М.С. // Российский вестник перинатологии и педиатрии – 2014 – №3.
Морфологическая характеристика синдрома Альпорта

Посудневская, А. И. Морфологическая характеристика синдрома Альпорта / А. И. Посудневская, А. Ю. Броницкая, К. В. Бондаренко. — Текст : непосредственный // Молодой ученый. — 2018. — № 13 (199). — С. 87-89. — URL: https://moluch.ru/archive/199/48981/ (дата обращения: 10.02.2022).
В данной статье отражены классические морфологические проявления синдрома Альпорта на примере данных 18 нефробиопсий детей от 4 до 18 лет с данной патологией.
Ключевые слова: гломерулонефрит, нефробиопсия, синдром Альпорта.
СиндромАльпорта (СА)— этиологически гетерогенное наследственное заболевание моногенной природы. Причина заболевания лежит в мутации одного из генов: COL4A5, COL4A4, COL4A3. При классическом варианте СА мутация происходит в гене COL4A5, расположенном на длинном плече Х-хромосомы (Хq22.2), который кодирует 5-цепь коллагена IV типа (НГ).
Распространенность СА составляет 1:5000. Он лежит в основе развития 1 % всех случаев хронической почечной недостаточности (ХПН) в Европе. 2,3 % случаев трансплантации почки проводится больным с СА [1].
Классические изменения почечной паренхимы при синдроме Альпорта представляют сочетание морфологических черт хронического гломерулонефрита, пиелонефрита и интерстициального нефрита, но с неполной картиной каждой из патологий. Почки уменьшены в размерах, их структура мелкозернистая или палисадная. На срезе коркового вещества часто имеются желтые линейные прожилки. Первичные изменения наблюдаются в гломерулярной мембране. Наиболее ранним поражением являются выраженная пролиферация эпителия в клубочках, интерстициальный фиброз или локальное расширение с атрофией канальцев, появляющиеся приблизительно одновременно. В большинстве случаев наблюдаются заполненные липидами пенистые клетки, не связанные с эпителиальными клетками канальцев. Эти пенистые клетки могут заполнять интерстициальную ткань, особенно в нижних отделах коры, располагаясь в виде рядов или гнезд. В большинстве случаев в интерстициальной ткани находят плазматические клетки, лимфоциты и отложения кальция. Канальцевые изменения включают атрофию эпителия и расширение некоторых канальцев. Иногда наблюдается хронический интерстициальный нефрит, при котором изначально мало клубочков.
При анализе нефробиопсий 18 пациентов на базе патологоанатомического бюро 3-ей городской детской клинической больницы г. Минска в большинстве случаев (83 %) обнаружен диффузный или фокальный, глобальный или сегментарный мезангиопролиферативный гломерулонефрит. У 17 % обследованных выявленные изменения расценены как фокально-сегментарный гломерулосклероз. Тубулоинтерстициальные изменения включали обнаруженные во всех случаях пенистые клетки, одиночные и сформировавшиеся в виде кластеров в интерстиции и канальцах, 39 % случаях — очаговый межуточный склероз с атрофией канальцев, в 28 % случаев — очаговое межуточное воспаление разной степени выраженности. При иммуногистохимии в большинстве случаев (61 %) экспрессия иммуноглобулинов A, G, M и С3 С1q фракций комплемента отсутствовала, в 33 % случаев выявлена экспрессия иммуноглобулина M, в 11 % случаев — экспрессия иммуноглобулина M и С3 С1q фракций комплемента. У 8 пациентов, которым проведено иммуногистохимическое исследование с применением антител к α3 и α5 субъединицам коллагена IV типа, выявлена гетерогенность иммуногистохимического окрашивания — от полного отсутствия до сохранения обеих субъединиц.
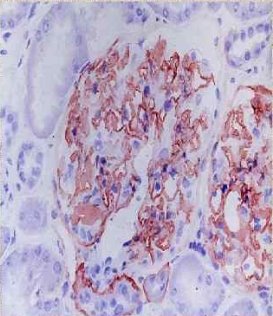
Рис. 1. Иммуногистохимическое исследование с применением антител к α3 (контроль)
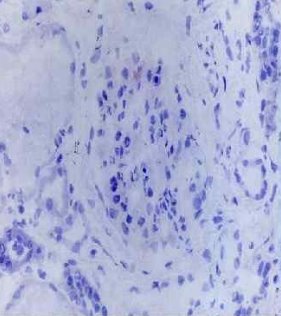
Рис. 2. Иммуногистохимическое исследование с применением антител к α3 (СА)
Таким образом, синдром Альпорта в большинстве случаев морфологически проявляется мезангиопролиферативным гломерулонефритом с наличием пенистых клеток и отсутствием экспрессии иммуноглобулинов A, G, M и С3 С1q фракций комплемента.
- Atkin C. L., Gregory V. S., Border W. A. Alport syndrome. In: R. W. Schrier, C. M. Cottschalk (eds.) Diseases of the Kidney. 4th ed.Boston: Little 1989; 233.
Основные термины (генерируются автоматически): большинство случаев, фракция комплемента, изменение, интерстициальная ткань, клетка, экспрессия иммуноглобулина.
Синдром Альпорта у детей: особенности развития, симптомы, методы диагностики и лечение
Синдром Альпорта у детей (иначе – заболевание тонких мембран) – редкая наследственная патология почек, характеризующаяся изменением выработки коллагеновых волокон IV типа, при этом меняется мембранная структура почечных клубочков с одновременным изменением структуры отделов внутреннего уха и глаз. По статистике, встречается 1 случай на 5000 человек. Примечательно, что синдром диагностируют у 1% больных с различными нефроурологическими патологиями и у 3% пациентов, ранее перенесших трансплантацию почки.
Причины и пути наследования

Механизм развития заболевания обусловлен дефектностью генетической единицы, отвечающей за структуру цепей коллагена IV типа. По типу наследования синдром классифицируют:
- на доминантный, или X-сцепленный. Заболевание обусловлено мутацией гена в женской половой хромосоме X. Такой путь наследования встречается в 85% всех клинических случаев;
- аутосомно-рецессивный. Обусловлен мутациями в генах второй порядковой хромосомной единицы, встречается в 10% всех случаев синдрома Альпорта;
- аутосомно-доминантный. Наследственный нефрит вызван мутацией генов во второй порядковой хромосоме, встречается в 1–2% случаев и напоминает течение аутосомно-рецессивного типа.
По мере прогрессирования болезни полностью разрушаются оболочки гломерул, почечных канальцев, внутренних глазных и ушных структур. Механизм развития патологического изменения почечных структур можно объяснить несколькими ключевыми факторами: генной мутацией, нарушением выработки и строения коллагена, разрушением канальцевых и гломерулярных почечных единиц, патологией почек.
Типичные признаки
Болезнь тонких базальных мембран редко диагностируется сразу после рождения или внутриутробно. Специфические симптомы наблюдаются спустя 3–4 года после рождения и выражаются в следующих состояниях:
- гематурический синдром. Одно из ранних проявлений болезни. Обычно выявляется лабораторно. У мальчиков микрогематурия отмечается еще до 1 года, но планово такой анализ на фоне общего здоровья не проводится. Ученые считают, что если до 10 лет ребенка гематурия не выявлялась ни разу, то синдром Альпорта можно исключать на 98%;
- белок в моче. Состояние протеинурии чаще отмечается у мальчиков с доминантным путем наследования мутации. Обычно белок в моче не выявляется или его появление носит эпизодический характер;
- артериальная гипертензия. Стойкое повышение артериального давления характеризуется прогрессированием почечной недостаточности. Чем ниже клиренс креатинина, тем интенсивнее развитие гипертензии;
- тугоухость. Нарушение слуха на фоне нефротического синдрома возникает не всегда. Известны случаи семейного синдрома Альпорта, когда у всех больных нет слуховых изменений. Ранняя стадия тугоухости определяется только при аудиометрии;
- лентиконус передний. Состояние характеризуется выпячиванием глазного хрусталика кнаружи, встречается у 20–25% больных с наследственным нефритом. Офтальмологическое заболевание постоянно прогрессирует, поэтому больные вынуждены часто менять очки;
- роговичная дистрофия. Истончение сетчатки глаза встречается редко. Почти 90% больных с дистрофией роговицы имеют X-сцепленный путь геномной мутации;
- лейомиоматоз бронхов и пищевода. У детей возникает ранняя дисфагия – нарушение глотания. Появляются и иные симптомы: загрудинные и эпигастральные боли, рвота, одышка, сухой кашель. Подтвердить лейомиоматоз можно с помощью МРТ или компьютерной томографии.
По мере взросления ребенка нарастают симптомы почечной недостаточности: недомогание, интоксикация, артериальная гипертензия, резкое увеличение креатинина и мочевины в составе крови, ацидоз.
Диагностика

Комплексное обследование детей с синдромом Альпорта включает сбор анамнеза жизни, наследственного и клинического анамнеза, изучение жалоб. Учитывая наследственную природу заболевания, значение имеют случаи почечной недостаточности у близких родственников. Для уточнения диагноза проводят ряд исследований:
- общеклинические лабораторные исследования;
- УЗИ почек и почечных структур, органов мочеполовой системы;
- рентгенконтрастные методы исследования (экскреторная урография);
- ДНК-тест;
- биопсия почек.
Диагностика требует дополнительной консультации нефролога, уролога, генетика, офтальмолога, кардиолога, отоларинголога. Заболевание дифференцируют от других геномных мутаций, в частности, от синдрома “кошачьего крика” – нарушения строения 5 хромосомы, когда типичные признаки выражаются в нарушении слуха и зрения еще в раннем возрасте.
Лечение
Заболевание неизлечимо, на причины генетических мутаций невозможно повлиять медикаментозно или хирургически. Терапия направлена на облегчение симптомов болезни. Обычно схема лечения включает следующие группы препаратов:
- мочегонные (петлевые, тиазидные, растительного происхождения);
- внутривенное введение физраствора для предупреждения обезвоживания;
- средства на основе глюконата кальция и глюкозы для восстановления минерального и электролитного баланса;
- антигипертензивная терапия при вторичной артериальной гипертензии (ингибиторы АПФ, блокаторы медленных кальциевых каналов);
- гормоны;
- железосодержащие препараты при выраженном гематурическом синдроме.
При развитии тяжелой почечной недостаточности встает вопрос о заместительной терапии. Лучшим способом заместительной терапии у детей является трансплантация почки. Пересадка органа позволяет вернуть прежний образ жизни, улучшить состояние больного.
Общие клинические рекомендации для больных наследственной нефропатией заключаются в соблюдении охранительного режима и диеты, регулярных прогулках на свежем воздухе, приеме витаминных комплексов. Все лечебные меры должны отвечать возрастным интересам ребенка.
Осложнения
Основными осложнениями при синдроме Альпорта у детей являются глухота, нарушение зрения и недостаточность функции почек. Последнее состояние провоцирует развитие вторичных осложнений, угрожает жизни пациентов. Прогноз при синдроме Альпорта у детей преимущественно благоприятный, если прогрессия крайне медленная, а симптомы заболевания не выражены. Если гематурия не сопровождается протеинурией, состояние пациента не осложнено, то важно соблюдать предупредительные лечебные меры. Учитывая генетическую природу заболевания, предупредить развитие патологии у детей невозможно.
Синдром Альпорта: причины, симптомы, классификация, лечение, прогнозы

Скачать
ENG

Скачать
РУС
DOI: 10.2215/CJN.00580116
РЕЗЮМЕ
Синдром Альпорта – наследственное заболевание, характеризующееся прогрессирующей почечной недостаточностью, потерей слуха и аномалиями глаз. Наследование Х-сцепленное (85%) или аутосомно-рецессивное (15%). Многие нефрологи считают, что при синдроме Альпорта заболевают преимущественно мужчины. Однако женщины при Х-сцепленных заболеваниях страдают в 2 раза чаще. У женщин заболевание, как правило, остается недиагностированным, но у 15-30% из них к 60 годам развивается почечная недостаточность, а к среднему возрасту часто отмечается потеря слуха. Половина их сыновей и дочерей также страдают данным заболеванием. Аутосомно-рецессивный вариант синдрома Альпорта встречается реже, но часто ошибочно принимается за Х-сцепленное заболевание. Рецессивный тип наследования подозревают, когда у женщины рано развивается почечная недостаточность или лентиконус. В их семьях могут быть близкородственные браки. Прогноз для других членов семьи очень отличается от Х-сцепленного заболевания. У представителей других поколений, включая родителей и потомков, заболевание отсутствует, и в среднем только один из четырех их сиблингов унаследует данную патологию. У всех женщин с синдромом Альпорта диагноз должен быть подтвержден с помощью генетического исследования (даже если функция почек у них в норме) из-за риска развития почечной недостаточности у них и рисков у их потомков. Вид мутации указывает на тип наследования и вероятность передачи заболевания их детям, кроме того, вид мутации характеризует почечный прогноз как при Х-сцепленном, так и при рецессивном типах наследования. У женщин с Х-сцепленным синдромом Альпорта необходимо как минимум ежегодно определять альбуминурию и измерять уровень артериального давления. Экспертное руководство по диагностике и лечению синдрома Альпорта ( Expert guidelines for the diagnosis and management of Alport syndrome ) при наличии альбуминурии рекомендует проводить терапию блокаторами ренин-ангиотензин-альдостероновой системы (РААС) (с адекватной контрацепцией из-за риска тератогенного действия ингибиторов ангиотензинпревращающего фермента) с целью торможения прогрессирования почечной недостаточности. Согласно современным рекомендациям, женщины с аутосомно-рецессивным синдромом Альпорта должны получать терапию блокаторами РААС с момента установления диагноза. Кроме того, эти женщины должны быть направлены на медико-генетическое консультирование и информированы о своих репродуктивных возможностях, также им необходим тщательный мониторинг во время беременности с целью выявления развития АГ.
КОММЕНТАРИИ
Синдром Альпорта – наследственное заболевание, характеризующееся прогрессирующей почечной недостаточностью, потерей слуха и аномалиями глаз, включая образование рубцов на роговице, лентиконус, истончение сетчатки и пигментный ретинит. Тип наследования – Х-сцепленный (85%) или аутосомно-рецессивный (15%). Подозрение о Х-сцепленном варианте заболевания возникает, если у мужчин в семье наблюдаются более тяжелые проявления, а болезнь проявляется не в каждом поколении. Об аутосомно-рецессивном типе наследования говорят, когда заболевание возникает только в одном поколении, в семье имеются близкородственные браки или у молодой женщины развивается ТПН и потеря слуха или лентиконус.
Х-сцепленный вариант синдрома Альпорта обусловлен мутациями в гене COL 4 A 5, кодирующем a 5 цепь коллагена IV типа. Аутосомно-рецессивное заболевание является результатом двух мутаций in trans (в разных хромосомах) в генах COL 4 A 3 или COL 4 A 4, которые кодируют a 3 и a 4 цепи коллагена IV типа соответственно. a 3, a 4 и a 5 цепи образуют гетеротример, который является основой базальных мембран гломерулярного фильтра, роговицы, капсулы хрусталика и сетчатки (подобным тканевым распределением объясняются клинические проявления).
При Х-сцепленном синдроме Альпорта в гене COL 4 A 5 описано более 2000 мутаций, и более 1000 в генах COL 4 A 3 и COL 4 A 4 при рецессивной форме.
Клинические проявления синдрома Альпорта у мужчин с Х-сцепленной формой и у мужчин и женщин с рецессивным вариантом заболевания идентичны и включают гематурию, протеинурию, ТПН, лентиконус, истончение сетчатки и ретинопатию. К редким проявлениям относят лейомиоматоз (опухоли мягких тканей пищевода, бронхов), аневризму аорты и гигантские разрывы сетчатки.
У женщин с Х-сцепленным вариантом синдрома Альпорта заболевание часто остается недиагностированным. Однако в среднем женщины заболевают в два раза чаще мужчин.
У больных женщин следует применять контрацепцию, чтобы избежать наступления беременности во время приема ингибиторов АПФ из-за риска тератогенного действия последних. Некоторые женщины с данным заболеванием принимали решение не заводить детей из-за риска рождения больного ребенка. Если семейная мутация известна, то теперь им может быть предложено проведение пренатальной диагностики или предимплантационной генетической диагностики, но эти процедуры требуют планирования беременности и являются дорогостоящими.
Во многих странах (в США, Великобритании, Франции, Германии, Израиле, Австралии, Нидерландах, Китае) существуют группы поддержки больных ( http :// alportsyndrome . org / connect / international – groups / ). У них есть сайты и блоги с доступом к квалифицированным врачам, которые могут дать консультацию по генетическим рискам, оптимальным подходам к лечению и редким клиническим осложнениям.
Жак ШАНАР (Pr. Jacques CHANARD)

