
Синдром Денди-Уокера: признаки, диагностика и лечение заболевания
В данной статье главной идеей является изучение клинико-лабораторных особенностей Аномалии Денди-Уокера (АДУ). В триаду симптомов входят так же гипоплазия червя мозга, расширение III желудочка с формированием кисты задней черепной ямки. Патология названа в честь учёных, впервые её описавших. Врач американского происхождения Уолтер Эдвард Денди, которого считают одним из основоположников нейрохирургии, в 1921 году впервые описал заболевание. Его труды были дополнены исследованиями Эрла Уокера. Авторы данной статьи рассказывают о причинах по которым развивается данная аномалия, патогенез и методы диагностики. Самым ранних методов диагностики является УЗИ, с помощью которого признаки аномалии можно диагностировать во втором триместре на 18-20 неделе. Изучение темы основано на клиническом случае пациента с аномалией Денди-Уокера находившегося на лечении в отделении хирургическом отделении КГП «ОДКБ». В статье приводятся: анамнезы жизни и заболевания пациента, его неврологический статус, результаты обследования. У пациента в момент рождения гидроцефалия отсутствует, но в раннем детском возрасте до 3 лет клиника гидроцефалии стала прогрессировать, что привело к соответствующим жалобам, запоздалому обращению за медицинской помощью и отсутствие динамического врачебного наблюдения за ребенком на уровне ПМСП.

1. Балычевцева И.В., гадецкая С.Г., Безуглова И.А., Атрощенкова Г.А., Стороженко Л.П., Турпаков А.В., Квач Л.Э., Горбунова О.В.. Клинический случай синдрома Денди — Уокера // Випадок i3 практики – 2011.№3.С136-138
2. Петрова Л.А., Розанов А.В., Барашнев Ю.И., Панов В.О..Синдром Денди—Уокера у новорожденных // Российский вестник перинатологии и педиатрии.- 2010.№1.С.25-29.
3. Проценко Е.В., Перетятко Л.П., Фатеева Н.В., Сарыева О.П..Патоморфология гидроцефалии, связанной с аномалиями развития водопровода мозга // Российский медико-биологический вестник имени академика И.П. Павлова- 2018.№3.С337-344
4. Трубникова Л.И., Азизова Р.Р., Таджиева В.Д., Жданова В.Ю., Измайлова Ф.А., Пигина Г.Р. Диагностическая значимость комплексного исследования врожденных пороков развития центральной нервной системы плода // Ульяновский медико-биологический журнал.-2012.№3.С22-60
5. Шерстнева О.В., Пренатальная ультразвуковая диагностика врожденных пороков развития центральной нервной системы // Медицинский совет – 2012. №1. С82-85
Введение. Пороки развития нервной системы суммарно занимают второе место в структуре аномалий развития после врожденной патологии сердечно-сосудистой системы, причем около 80 % этих заболеваний представлены гидроцефалией различного генеза.
Аномалия Денди-Уокера (АДУ) объединяет собой триаду симптомов: гипоплазия червя мозга, расширение III желудочка с формированием кисты задней черепной ямки и внутренняя гидроцефалия.[1].
АДУ – врожденная патология с частотой встречаемости 1 случая на 10000-30000 новорожденных. К причинам приводящим к развитию АДУ относятся:внутриутробное инфицирование вирусами герпеса, краснухи, кори, цитомегаловируса, а также сахарный диабет, метаболические расстройства, ионизирующее излучение, прием беременной антибиотиков, внутриутробные травмы. Нельзя исключить и наследственную предрасположенность, из-за дефекта генов может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.[2].
Патогенез АДУ до конца не изучен. Существует две версии патогенеза. Первая предложена У. Денди и К. Блекфаном, о частичной или полной окклюзии отверстий Люшки и Мажанди. Отверстия Мажанди, соединяет III и IVжелудочки с большой цистерной головного мозга, в то время как отверстия Люшка, являются сообщением между желудочками и подпаутинным пространством оболочек мозга. Как следствие атрезии данных отверстий происходит патологическое накопление ликвора в желудочках мозга, увеличивается задняя черепная ямка, растет внутричерепное давление, образуется ликворная киста в задней ямке черепа, а так же обструктивная гидроцефалия. Которая является осложнением АДУ, а не проявлением синдрома. Другая версия предложена Е. Гарднером, о нарушении баланса в продукции спинномозговой жидкости в желудочках мозга, приводящем к сдавлению червя мозжечка, его вторичной гипоплазии. Возможна и первичная агенезия червя мозжечка как результат нарушения слияния ромбических губ на ранних стадиях органогенеза.Наличие дефекта червя мозжечка, приводит к увеличению размеров IV желудочка, создаются условия для его пролабирования в подпаутинное пространство и формирования кисты в области большой цистерны.[3].
Выделяют две формы аномалии Денди-Уокера: полную и неполную. Полная характеризуется полной агенезией червя мозжечка и наличием сообщения между IV желудочком и кистой в области большой цистерны. При неполной форме выявляется частичная агенезия червя мозжечка, в связи с чем коммуникация IV желудочка с кистой большой цистерны прослеживается не на всем протяжении червя.
Вариантами АДУ является открытый и закрытый тип. Отличие в наличии или отсутствии окклюзии отверстий Люшке и Мажанди и сообщением желудочка с подпаутинным пространством. Таким образом, две теории патогенеза имеют место быть. Так как в настоящее время, отсутствуют методы дородовой оценки проходимости этих структур головного мозга эти варианты не имеют клинического значения.
Пренатальная диагностика АДУ возможна во второй триместр беременности на 18-22 недели при ультразвуковом исследовании(УЗИ). УЗ-признаки: размеры IV желудочка значительно увеличены, недоразвитие мозжечка, наличие в черепной ямке полости, заполненной жидкостью(кисты). Визуализация червя мозжечка при трансабдоминальном УЗИ возможна уже с 15-16 недель в сагиттальной плоскости и с 20 недели – в горизонтальной. По данным литературы ранняя пренатальная диагностика АДУ была осуществлена при трансвагинальной эхографии в 12-14 недель беременности. При подозрении на АДУ или наследственной отягощенности рекомендуется выполнять кариотипирование.
Развитие АДУ может быть, как постепенным, так и быстрым. В постнатальном периоде обращение на себя внимание медленное моторное развитие ребенка и прогрессирующее расширение черепа, истончение и выпячивание кости в затылочной части черепа. В раннем детском возрасте проявляется симптомами внутричерепной гипертензии, то есть раздражительность, тошнота, судорожный синдром, нарушение зрения будут характеризовать данное состояние, а также наличием мозжечковой симптоматики (статическая атаксия, нарушение координации движений, нистагм). В 60-75% АДУсочетается с другими аномалиями головного мозга, а также с пороками развития органов и систем плода аномалиями развития сердца, палатосхизом, полидактилией. Развитие умственной отсталости развивается в следствие агенезией мозолистого тела и нарушением миграции нейронов в процессе созревания больших полушарий. Неврологическая симптоматика обуславливает тяжесть состояния. Положение больного с АДУ в постели становится вынужденным, на боку с запрокинутой головой, возможен, крик «мозговой», монотонный, плавающие движения глазных яблок, симптом «заходящего солнца», горизонтальный нистагм. Реакция на звук и свет может отсутствовать. Рефлексы периода новорожденности угнетены.Гиподинамия, спонтанная двигательная активность снижена. Мышечный тонус в конечностях дистоничный, тетрапарез. Физическое развитие негармоничное, часто ребенок пониженного питания, тургор тканей снижен.
Постнатальная диагностики, у детей до 1 года основана на нейросонографии, которая позволяет визуализировать кисту задней черепной ямки. Специфичным методом при АДУ является МРТ и КТ исследования головного мозга пациента.[3,4]
При диагностики повышенного внутричерепного давления(ВЧД) информативна рентгенография черепа. У детей признаки повышения ВЧД проявляются расширением черепных швов зубцы швов удлиняются, расширяются венечный, стреловидный и иногда ламбдовидный швы. У маленьких детей резко задерживается закрытие родничков. При выраженной гипертензии у детей изменяется конфигурация черепа, основание продавливается вниз (особенно средняя черепная яма), расширяются черепные отверстия, истончаются кости свода, изменяется рельеф их внутренней поверхности. Резко изменяется конфигурация турецкого седла. Череп приобретает форму шара, объем его увеличивается, что несколько компенсирует гипертензию.
Лечение: проводится оперативное шунтирование четвертого мозгового желудочка для снижения внутричерепного давления, за счет оттока ликвора. Для коррекции гипертонуса мышц и двигательных нарушений используются препараты и физиотерапевтические методы (гимнастика, массаж). Умственная отсталость практически не поддается терапии.
Целью нашего исследования является изучение клинико-лабораторных особенностей Аномалии Денди-Уокера.
Материалы и методы: Нами описан клинический случай пациента С., в возрасте 5 лет, с аномалией Денди-Уокера, находившегося на лечении в отделении КГП «ОДКБ». Данному ребенку были проведены комплексное обследование, консультация узких специалистов, оперативное лечение.
Приводим клинический случай
Пациент С. 5 лет. Рост 117 см, вес 18,5 кг. С диагнозом: Врожденный порок развития (ВПР) центральной нервной системы (ЦНС). Аномалия Денди Уокера. Внутренняя прогрессирующая окклюзионная гидроцефалия в стадии декомпенсации. Мозжечковая атаксия. Хронический гастродуоденит в стадии ремиссии. ДЖВП по смешанному типу. Гиперметропический астигматизм прямого типа. Остаточное сходящее косоглазие
Anamnesis vitae: Ребенок от 2 беременности, 1 родов. Вес при рождении 3710,0 гр., рост 57см, окружность головы при рождении 33см. Беременность протекала с угрозой прерывания беременности, в первом триместре на фоне ОРВИ. Роды срочные, физиологичные, в сроке 39 недель+5 дней. Состоял на «Д» учете у невропатолога с диагнозом: Гипоксия до 1 года. Состоит на «Д» учете у окулиста с диагнозом: Остаточное постоянное сходящееся левостороннее неаккомадационное альтернирующее при ковер – тесте косоглазие с вертикальным компонентом. Амблиопия.
Перенесенные заболевания: ОРВИ. Наследственность по линии матери – Сахарный диабет у бабушки 1 типа, Артериальная гипертензия. В ноябре 2018 года проведен 2 этап операции на глаза. Аллергологический анамнез не отягощен.
Anamnesis morbi: Динамику окружности головы (ОГ) не проводили. В возрасте 4 лет, впервые было сделано МРТ головного мозга, в связи с жалобами на частые головные боли, а также жалоб на увеличение окружности головы.
МРТ головного мозга от 20.11.2018г. МР – признаки наиболее характерные для аномалии Денди – Уокера. Внутренней гидроцефалии. Энцефалопатии смешанного генеза.
На рентгенографии черепа (2 проекции) от 16.11.2018г. выраженная черепно-мозговая гипертензия.
В ноябре 2018 года консультирован нейрохирургом, на момент осмотра, оперативное лечение не было показано.
04.01.2019г. повторно консультирован нейрохирургом. Заключение: ВПР. ЦНС. Внутренняя гидроцефалия в стадии компенсации; на момент осмотра оперативное лечение не показано. Консультирован невропатологом, диагноз: Резидуальная энцефалопатия. Гипертензионный синдром. Аномалия Денди-Уокера. Назначено лечение: Пантокальцин в дозировке 250 мг., Магний В6, Ноофен. В динамике без улучшения.
ЯМРТ от 27.03.2019г.: внутренняя гидроцефалия. В сравнении с 19.11.2018г -отрицательная динамика, за счет расширения боковых желудочков.
Дано направление на оперативное лечение в ОДКБ.
Status praesens: Состояние при поступлении в стационар: Средней степени тяжести, тяжесть обусловлена поражением ЦНС. На осмотр реагирует беспокойством. Кожные покровы бледноватые, чистые от сыпи. Слизистые розовые, влажные. Подкожно-жировой слой удовлетворительный. Периферические лимфоузлы не увеличены. Костно-суставная система без деформации. Носовое дыхание свободное. В легких дыхание проводится по всем полям, пуэрильное, хрипов нет. Область сердца визуально не изменена, сердечные тоны ясные, ритмичные. Живот правильной формы, симметричен, не вздут, при пальпации мягкий, безболезненный. Печень и селезенка не увеличена. Мочеиспускание свободное, безболезненное. Стул был 1 раз, оформленный.
Неврологический статус: сознание ясное, ОГ=54.5 см. ЧМН: Зрачки ОS=OD, фотореакция сохранена. Взгляд не фиксирует. Сухожильные рефлексы вызываются слабо, нижних конечностях. Д=С. мышечный тонус в нижних конечностях снижен, кисти паретичны. Повороты головы в обе стороны. Менингеальные симптомы отрицательные.
30.03.2019 проведена вентрикулостомия. Операцию перенес успешно. Выписан на 10 день.
Неврологический статус при выписке: сознание ясное, ОГ=54.0см. Черепно-мозговые нервы: Зрачки ОS=OD, фотореакция сохранена. Взгляд фиксирует. Сухожильные рефлексы вызываютсяD=S. Мышечный тонус в нижних конечностях с положительной динамикой. Самостоятельно ходит. Выполняет пальценосовую пробу. Повороты головы в обе стороны. Менингеальные симптомы отрицательные.
Результаты и обсуждения: По мнению большинства авторов, гидроцефалия у большинства детей с пороком Денди — Уокера в момент рождения отсутствует и развивается на протяжении первых месяцев жизни [2], что совпадает с данными нашего пациента. В 1 год пациент снят с диспансерного учета невропатологом, следовательно, симптоматика была компенсирована. В раннем детском возрасте до 3 лет клиника гидроцефалии стала прогрессировать, что привело к соответствующим жалобам, запоздалому обращению за медицинской помощью и отсутствие динамического врачебного наблюдения за ребенком на уровне ПМСП.
В 65 % случаев, по данным R. McLaurin (1985), порок сочетается с другими аномалиями головного мозга — агенезией мозолистого тела, энцефалоцеле, полимикрогирией, агирией, гетеротопией серого вещества, а также с поражениями других органов и систем (полидактилией, синдактилией, врожденными пороками сердца, поликистозом почек, расщелинами неба и др.). У нашего пациента видимых пороков развития и стигм дизэмбриогенеза не обнаружено, что свидетельствует о более благоприятном прогнозе для жизни. С целью выявления наследственной отягощенности рекомендуется выполнить кариотипирование данному ребенку.
Прогноз для жизни и здоровья при синдроме Денди — Уокера зависит от наличия сочетанных аномалий развития, хромосомных аномалий и срока диагностики варьирует от практически нормального постнатального развития до тяжелой инвалидности и даже гибели ребенка [4,5]. У нашего пациента необходимо отметить тяжелое поражение мозга и исходом в инвалидность, грубую задержку темпов психомоторного и физического развития.
По данным литературы, показатели постнатальной заболеваемости и смертности выше в тех случаях, когда синдром диагностирован в пренатальном периоде, а не у новорожденного.
Выводы: Пренатальная диагностика гидроцефалии и детальное клинико-лабораторное обследование новорожденных с признаками начинающейся гидроцефалии — залог ранней диагностики синдрома Денди—Уокера, оказания ребенку своевременной медикаментозной помощи, а в дальнейшем – возможной нейрохирургической коррекции врожденного дефекта.
Синдром Денди-Уокера
Синдром Денди-Уокера – это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.
МКБ-10
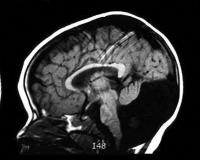


Общие сведения
Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.
Причины
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Патогенез
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Классификация
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
В 1989 г. американским педиатром и нейрорадиологом Джеймсом Барковичем была предложена альтернативная классификация, базирующаяся на результатах МР-диагностики. Выделена группа заболеваний, названная «комплексом Денди-Уокера», который включает 4 клинических формы: классическую аномалию (полная форма), вариант Денди-Уокера (менее грубые нарушения), кисту кармана Блейка и mega cisterna magna.
Симптомы
Основное проявление синдрома Денди-Уокера – гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера – симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
Осложнения
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Диагностика
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
Дифференциальная диагностика
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
Хирургическое лечение
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Прогноз и профилактика
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. – 2018. – №2.
2. Аномалия Денди-Уокера – редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. – 2017. – №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. – 2010. – №1.
Синдром Денди-Уокера: понятие, симптомы, диагностика, лечение
Синдром Денди-Уокера – тяжелый порок развития головного мозга. Эта врожденная аномалия у детей характеризуется недоразвитием или гипоплазией червя мозжечка, кистозным поражением четвертого желудочка и расширением задней черепной ямки.
Синдром Денди-Уокера – сложный симптомокомплекс, обусловленный врожденным пороком головного мозга, сформированным в процессе эмбриогенеза и отличающимся тяжелым течением. В основе патологии лежит одновременное поражение важнейших мозговых структур: мозжечка, желудочков, продолговатого мозга, черепно-мозговых нервов и крупных сосудистых стволов. Гипоплазия мозжечка характеризуется уменьшением его размеров и снижением функции. Возможно его полное отсутствие. Расширение желудочков мозга обусловлено врожденными кистами или опухолями. Эти признаки патологии всегда сочетается с гидроцефалическими изменениями, которые являются не проявлениями, а осложнениями синдрома.

Неполноценность ликворных путей приводит к скоплению цереброспинальной жидкости в желудочках, которая в норме должна выводится из головного мозга. Ее патологическое скопление способствует развитию водянки мозга, проявляющейся признаками гипертензионно-гидроцефального синдрома. Благодаря постоянному давлению ликвора начинается расширение костей черепа. Синдром Денди-Уокера сопровождается множественными пороками внутренних органов: сердца, сосудов, почек, костей.
Синдром открыли хирурги из Америки — Денди в 1921 году и Уокер в 1944 году. Недуг регистрируется у девочек несколько чаще, чем у мальчиков. У новорожденных быстро расширяется черепная коробка, замедляется психомоторное развитие, появляются признаки гидроцефального синдрома, внутричерепной гипертензии, поражения мозжечка и черепно-мозговых нервов. Практически у каждого больного имеются сопутствующие аномалии: гипоплазия мозолистого тела, врожденные пороки сердца, атипичное строение лицевого скелета.
Патологию можно диагностировать еще на этапе беременности. Эхографические признаки недуга специалисты обычно обнаруживают во втором триместре при проведении планового УЗИ. По мере развития плода УЗИ-показатели становятся более выраженными. В пользу синдрома Денди-Уокера свидетельствуют следующие признаки: кисты мозга, гипотрофия мозжечка, вентрикуломегалия. Аномальное развитие плода, обнаруженное с помощью УЗИ — повод для искусственного прерывания беременности.
Этиология и патогенез
Причины синдрома в настоящее время остаются неизвестными. Существует несколько врачебных предположений относительно этиологических факторов недуга. Одна из теорий повествует о первичном недоразвитии структур головного мозга, другая — о повышенной секреции ликвора в процессе эмбриогенеза. Все остальные этиопатогенетические гипотезы менее популярны и неправдоподобны.

Формирование мальформации Денди-Уокера связывают с наличием следующих патологий:
- Сирингомиелии,
- Отсутствием мозолистого тела,
- Патологии мозжечка,
- Заращения или окклюзии ликворных каналов,
- Аномального увеличения желудочков мозга,
- Кист и опухолей.
Сдавление структур мозга приводит к неврологической дисфункции. Череп увеличивается в окципитальной и фронтальной плоскости. В первые два месяца жизни окружность головы растет наиболее интенсивно. У новорожденных наблюдается расхождение швов на черепе и истончение его костей.
К факторам, способствующим развитию синдрома, относится негативное воздействие внешней среды на организм беременной женщины, а также имеющиеся хронические заболевания. Наиболее распространенные провоцирующие факторы:
- Вирусная инфекция — герпес-вирусы, краснуха, корь, цитомегаловирус,
- Вредные привычки,
- Наркомания,
- Сахарный диабет и другие метаболические расстройства,
- Аутоиммунные заболевания,
- Ионизирующее излучение,
- Прием антибиотиков,
- Внутриутробные травмы.
Наиболее опасными эти факторы являются в первом триместре беременности, когда происходит активное развитие ЦНС плода. Во всех остальных случаях причиной синдрома становится наследственная предрасположенность. При наличии синдрома у одного из родителей риск появления его у ребенка очень высок.
Патогенетические звенья аномалии Денди-Уокера:
- Нарушение развития ликвороотводящих путей,
- Патологическое накопление спинно-мозговой жидкости в желудочках мозга,
- Сдавление ликвором участков мозга,
- Внутричерепная гипертензия,
- Поражение ядер черепно-мозговых нервов,
- Появлению клинической симптоматики,
- Образование в мозге кистозных полостей, содержащих ликвор,
- Увеличение полости в размерах,
- Деформация черепа,
- Еще большее сдавление структур головного мозга,
- Нарушение полноценного развития мозга,
- Утрата функциональности органа.
Симптоматика

Признаки мальформации у плода, выявляемые с помощью УЗИ:
- Кисты и опухоли в головном мозге,
- Уменьшение размеров мозжечка и снижение его функций,
- Вентрикуломегалия,
- Внешние уродства — незаращение неба и верхней губы,
- Окклюзия отверстий и отсутствие каналов в головном мозге,
- Почечные аномалии,
- Неполноценность структур ликвороотводящей системы,
- Отмирание полушарий мозжечка,
- Свищи в мозге.
Большинство ученых признают синдром Денди-Уокера врожденной аномалией. Но несмотря на это, клинические признаки патологии впервые могут проявиться у маленького ребенка, школьника, подростка и даже взрослого человека.

Симптомы патологии у новорожденных:
- Непропорционально большая голова по сравнению с туловищем,
- Выпячивание затылка,
- Остеомаляция костей черепа,
- Выбухание родничка,
- Ограничение или снижение двигательной активности рук и ног,
- Непроизвольные движения конечностей,
- Снижение сопротивления и упругости при сдавливании кожи и мягких тканей,
- Аритмия,
- Постоянный плач, капризность, беспокойство ребенка,
- Слабый крик,
- Деформация лица,
- Гиперрефлексия,
- Повышенная возбудимость,
- Подергивания глазных яблок,
- Косоглазие,
- Судорожное сокращение мышц конечностей,
- Постоянное сгибание рук и ног, обусловленное напряженностью мышц,
- Кратковременная остановка дыхания во сне,
- Поражение лицевого нерва.
Клиника синдрома Денди-Уокера у взрослых складывается из следующих признаков:
- Мозжечковая симптоматика – нистагм, неточные и широкие движения, головокружение, изменение походки, дизартрия, скандированная речь, тремор, дискоординация движений, невозможность выполнять быстрые чередующиеся действия;
- Признаки высокого внутричерепного давления и гидроцефалии — беспокойство, тошнота, рвота, тупая, ноющая головная боль по утрам, головокружение, бледность кожи, слабость, вялость, реакция на яркий свет и громкий звук, хождение «на цыпочках» при гипертонусе мышц ног, заторможенность, мнестические расстройства, невнимательность, судорожный синдром, коматозное состояние;
- Нарушение двигательной координации – трудности при ходьбе, мышечный гипертонус, спазмы и напряженность мускулатуры, частые судорожные припадки;
- Неизлечимая отсталость в интеллектуальном развитии – больные не узнают родных, не могут читать, отличать буквы, писать, становятся раздражительными, забывчивыми;
- Симптомы аномалии почек — периодическая боль в пояснице, повышение артериального давления, отеки на лице вокруг глаз, на теле и ногах, трудности с мочеиспусканием, жжение и зуд при этом процессе, мутная моча с кровяными выделениями;
- Признаки врожденного порока сердца – одышка, возникающая на фоне незначительной физической нагрузки, усиление сердцебиения, проявление общей слабости и бледности кожи, цианоз, боль в сердце, обмороки, отечность конечностей;
- Синдактилия, деформация лица и всего черепа, увеличение объема головы;
- Нарушение зрительной функции – снижение остроты зрения, диплопия.
Исход синдрома во многом зависит от глубины поражения ЦНС, а также от наличия сочетанной патологии, сопровождающей данный недуг. К серьезным осложнениям заболевания относятся интеллектуально-психологические пороки, замедление психомоторного развития, невротические болезни. Недостаточность высших психических функций, когнитивные расстройства и двигательная неловкость – самые распространенные последствия заболевания.
Видео: пример взрослого пациента с синдромом Денди-Уокера (eng)
Диагностирование
Диагностика синдрома бывает пренатальной и постнатальной. В первом случае аномалию обнаруживают у плода во время УЗИ беременной женщины. Родители должны принять решение о прерывании беременности или сохранении жизни малыша, который будет только мучиться и страдать.

синдром Денди-Уокера на диагностическом снимке
После появления больного ребенка на свет его осматривают и выявляют характерные симптомы, которые видны невооруженным глазом: мышечный гипертонус, размашистость движений, увеличенный объем головы. Затем специалисты собирают семейный анамнез, опрашивая родителей о наличии в семье подобных заболеваний. Определение неврологического статуса – наличие нистагма, дискоординации движений и прочих признаков аномалии.
Чтобы обнаружить пороки внутренних органов, необходимо пройти дополнительную инструментальную диагностику: УЗИ сердца, ангиография, КТ и МРТ мозга, почек. Больным показано офтальмологическое обследование, эхокардиография и другие необходимые методы исследования жизненно важных функций организма. Медико-генетической консультирование с изучением кариотипа проводится лицам, имеющим в роду наследственные заболевания.
Лечебный процесс
Синдром Денди-Уокера с трудом поддается лечению. Специалисты назначают больным консервативную терапию, направленную на устранение признаков гидроцефалии, пороков развития сердца и почек.

Лекарственное лечение проводится при наличии легкой формы синдрома с минимумом клинических проявлений. Пациентам назначают мочегонные средства «Фуросемид», «Верошпирон», «Индапамид», успокоительные препараты «Грандаксин», «Сибазон», обезболивающие медикаменты «Ибупрофен», «Кетарол», миорелаксанты «Мидокалм», «Сирдалуд».
Успокоительные отвары снимают раздражительность и избавляют больного от цефалгии. С целью коррекции мышечного гипертонуса и двигательной дисфункции назначают физиотерапевтические процедуры: расслабляющие ванны с настоем мяты, мелиссы, валерианы или эфирного масла хвои; глубоко прогревающие методики – озокерит и парафин; электрофорез с лекарственными средствами на очаг поражения; а также ЛФК и лечебный массаж.

Оперативное вмешательство показано при челюстно-лицевых аномалиях. Для снижения внутричерепного давления проводят шунтирующие операции или вентрикулостомию. Они улучшают ликвороотток, тем самым уменьшая выраженность проявлений гидроцефального и гипертензионного синдромов. Шунтирование восстанавливает работу мозга, избавляет от неврологических расстройств и продлевает больному жизнь. При этом процент ранней смертности снижается в разы.
В четвертый желудочек мозга вводят шунт, по которому избыток ликвора вытекает в брюшную или грудную полость. Специальные дренажные трубки оснащены клапанным аппаратом, препятствующим обратному ток ликворной жидкости. Подобное лечение значительно облегчает состояние больных. При этом исчезают лишь клинические проявления болезни. Шунтирующие операции, несмотря на свою надежность и функциональность, не способны устранить причину синдрома.
Профилактика и прогноз
Врожденные синдромы, прогрессирующие на генетическом уровне, невозможно предупредить. Профилактические мероприятия проводить бессмысленно. Специалисты рекомендуют всем беременным женщинам регулярно посещать гинеколога, проходить УЗИ в каждом триместре и строго соблюдать рекомендации лечащего врача.
Риск развития патологии у плода повышается, если будущая мать курит, употребляет алкоголь, ведет нездоровый образ жизни. Последствие подобного поведения — образование у плода пороков внутренних органов.
Прогноз синдрома в целом неблагоприятный. Большинство больных детей с выраженными признаками патологии погибают на первом году жизни. Остальные малыши не смогут жить полноценно. Из-за имеющихся двигательных и психических нарушений они не сидят и не стоят самостоятельно, растут умственно отсталыми. Работа с психологами, дефектолога и прочими специалистами, как не помогает таким пациентам. Избавиться от отклонений в психофизическом развитии и вылечить неврологические дефекты полностью невозможно. Больные дети нуждаются в тщательном уходе и требуют особого внимания. Врожденные пороки ЦНС делают человека инвалидом. Мозговая дисфункция и прогрессирующая гипертензия – причины гибели больного.
Синдром Денди-Уокера — неизлечимое врожденное заболевание, характеризующееся аномалией головного мозга, дисфункцией мозжечка и ликвородинамическими расстройствами. Подобные патологические изменения сопровождаются ярко выраженной клинической симптоматикой.
Синдром Денди-Уокера: понятие, причины, формы, лечение, прогноз
Аномалии формирования центральной нервной системы — отнюдь не редкость. Эта патология занимает второе место по частоте после врожденных дефектов развития сердца и сосудов и в большинстве случаев представлена гидроцефалией самого разного происхождения. Помимо тяжелых нарушений, сопровождающих патологическое развития мозга, пороки ЦНС несут высокий риск смертельного исхода, а по данным статистики они лидируют по числу смертей в младенческом возрасте.
Синдром Денди-Уокера — одна из разновидностей нарушения формирования головного мозга еще во время внутриутробного развития. И хотя частота его относительно невелика (всего 1 случай на 25-30 тысяч младенцев), диагностируется порок едва ли не у каждого десятого малыша с врожденной гидроцефалией, которая и служит одним из основных проявлений патологии.
Впервые синдром был описан почти столетие назад, чуть позже — предложен метод хирургической коррекции, однако по прошествии многих десятилетий патология так и остается неизлечимой, и даже современная медицина не в состоянии исправить тяжелые последствия нарушений эмбриогенеза, возникающие при аномалиях головного мозга.
Синдром Денди-Уокера — это порок задней черепной ямки, при этом основные структурные изменения касаются мозжечка, ликвороотводящих путей, четвертого желудочка мозга. Аномалия диагностируется во время беременности посредством ультразвукового осмотра, после чего женщине может быть предложено прерывание беременности по медицинским показаниям.

Конечно, любые отклонения в развитии плода — это всегда большой стресс и переживания для родителей, однако в случае врожденного порока мозга надеяться на чудо не приходится — прогноз серьезный, а смертность высока. Малыши с сочетанными пороками мозга и других органов погибают в раннем возрасте как от мозговой дисфункции, так и от присоединившейся инфекции.
Аномалия Денди-Уокера часто сочетается с другими нарушениями и генетическими заболеваниями, зачастую несовместимыми с жизнью — микроцефалия (недоразвитие полушарий мозга), мозговые грыжи. У младенца может быть диагностирован генетически обусловленный поликистоз почек, недоразвитие зрительных нервов и глазных яблок со слепотой, аномалии сердечно-сосудистой системы.
Все эти неблагоприятные факторы, возможность сочетанной патологии многих органов делают синдром Денди-Уокера серьезнейшей проблемой в случае, если малышу дадут возможность родиться. Лечение патологии, как правило, симптоматическое, направленное на поддержание главных систем жизнеобеспечения и борьбу с инфекционными осложнениями. В редких случаях применяют хирургическую операцию, которая лишь облегчает явления гидроцефалии, но не ликвидирует ее полностью.
Почему возникает синдром Денди-Уокера?

Причины аномалий развития задней черепной ямки до сих пор не выяснены, однако выделен ряд факторов, способствующих подобным врожденным порокам:
- Инфицирование во время беременности цитомегаловирусом, перенесенная краснуха;
- Употребление алкоголя, курение, наркомания во время беременности;
- Экстрагенитальная патология, особенно — сахарный диабет у будущей мамы.
Под действием перечисленных причин или среди полного благополучия может возникнуть спонтанная мутация в генах, предрасполагающая к нарушению развития мозга. Особенно высок риск пороков при действии неблагоприятных факторов во время первого триместра гестации, когда и происходит закладка основных структур центральной нервной системы.
В части случаев синдром Денди-Уокера носит наследственно-обусловленный характер, то есть возникает из-за дефекта генов и может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.
Что происходит с мозгом при синдроме Денди-Уокера?
Анатомически классический вариант мальформации задней черепной ямки включает:
- Гидроцефальный синдром разной степени выраженности;
- Кистозную полость в задней части черепа с расширенным четвертым желудочком мозга;
- Отсутствие или недоразвитие червя мозжечка, недоразвитие его полушарий.
Червь мозжечка — это структура, расположенная между его половинами и несущая в себе проводящие нервные волокна. При аномалии Денди-Уокера он может быть представлен небольшой щелью или широким пространством между гемисферами органа. При неполном отсутствии червя щелевидное расширение образуется лишь в нижней его части. На фоне патологии этого отдела наблюдается недостаточное развитие и мозжечковых полушарий.
Именно дефект мозжечкового червя в виде расщелины считается характерным признаком аномалии Денди-Уокера, позволяющим отличать ее от недоразвития на фоне других пороков мозга.

Обязательный компонент синдрома — кистозная полость в задней части черепа, которая сообщается с желудочковой системой или замкнута и происходит из четвертого желудочка. Избыток ликвора и гидроцефальный синдром связаны с недоразвитием отверстий этого отдела мозга (Люшка и Мажанди), обеспечивающих отток жидкости. При частичном сообщении желудочковой кисты с субарахноидальным пространством последнюю назовут открытой, в противном случае киста считается закрытой.
Киста четвертого мозгового желудочка может самопроизвольно вскрыться в 3-ий желудочек или субарахноидальное пространство. В этом случае симптомы окклюзии ликворных путей несколько ослабнут. Выраженность гидроцефального синдрома вариабельна — от небольшого расширения желудочковой системы до высокой степени окклюзионной гидроцефалии с отсутствием возможности для циркуляции ликвора.
Многие специалисты отмечают, что у большинства малышей с синдромом Денди-Уокера при рождении гидроцефалии как таковой нет, а формируется она и прогрессивно нарастает в течение первых нескольких месяцев жизни, поэтому факт отсутствия гидроцефального синдрома сразу после родов при наличии диагностированной внутриутробно патологии не является поводом для пересмотра диагноза и необоснованных надежд, с этим связанных.
Более, чем в половине случаев синдрома Денди-Уокера у детей помимо описанных структурных аномалий обнаруживаются и другие дефекты мозга — недоразвитие или отсутствие мозолистого тела, мозговые кисты, недоразвитие или отсутствие извилин, смещения серого вещества относительно его правильной локализации, что еще больше усугубляет течение и без того тяжелой патологии.
По данным МРТ было выделено несколько разновидностей синдрома Денди-Уокера:
- Классический тип аномалии — задняя черепная ямка расширена, четвертый желудочек кистозно изменен, червь мозжечка частично или полностью недоразвит, полушария его гипоплазированы, а намет находится выше, чем в норме, желудочковая система не сообщается с подпаутинным пространством, часто наблюдаются мозговые кисты и отсутствие мозолистого тела, практически у всех пациентов есть гидроцефалия, возможно сдавление стволовых структур. Порок проявляется клинически уже с рождения и имеет неблагоприятный прогноз.
- Вариант Денди-Уокера — морфологические признаки выражены меньше, чем при классической форме, гипоплазирован нижний отдел червя мозжечка, желудочки сообщаются с кистой и ликворными пространствами, обеспечивая отток ликвора, поэтому гидроцефалия наблюдается редко. Задняя черепная ямка имеет нормальные размеры, стволовые структуры не сдавливаются.
- Киста кармана Блейка — расширение желудочковой системы с гидроцефальным синдромом, киста расположена под или за мозжечком, червь развит относительно хорошо. Четвертый желудочек расширен, но не сообщается с затылочной ликворной цистерной.
- Mega cisterna magna — вариант очагового расширения подпаутинного пространства в задней и нижней частях задней ямки черепа с увеличением объема затылочной цистерны, которая сообщается с четвертым желудочком и субарахноидальным пространством.
Проявления заболевания
Симптоматика синдрома Денди-Уокера разнообразна. Возможны как практически нормальное развитие ребенка после рождения, так и грубые неврологические изменения, влекущие тяжелую инвалидность и даже смерть. По некоторым данным, нормальное развитие интеллекта бывает в половине случаев изолированного порока, возможно даже случайное обнаружение синдрома при обследовании взрослых.

дети с синдромом Денди-Уокера
Внутриутробное течение патологии определяется степенью поражения мозга, нарастанием гидроцефалии, наличием других пороков развития. Прогноз значительно хуже при диагностике синдрома до рождения. При глубоких нарушениях в формировании мозга на первый план среди других проявлений выступает гидроцефальный синдром:
- Увеличение диаметра головы;
- Выбухание родничка.
Увеличение диаметра черепа происходит, главным образом, за счет затылочной области, в которой образуется киста, вызывающая истончение и растяжение костной основы. При выраженной гидроцефалии голова малыша активно растет на протяжении первых двух месяцев, параллельно происходит расхождение швов между костями спереди или в заднем отделе. Кроме того, характерны:
- Повышение нервной возбудимости (рефлексов);
- Глазодвигательные расстройства — нистагм, косоглазие;
- Приступы остановки дыхания;
- Парез лицевого нерва.
Симптомы мозжечковых нарушений у новорожденных выявить невозможно, и даже тяжелый дефект формирования мозжечковых структур далеко не всегда вызывает значимые признаки атаксии (двигательных расстройств), которая регистрируется всего у трети пациентов.
Значительно чаще, нежели двигательные нарушения, возникают расстройства психической деятельности, интеллекта, которые проявляются на фоне общей двигательной «неловкости». У 25% больных с гипоплазированным мозжечком имеются признаки аутизма, в связи с чем специалисты пытаются найти взаимосвязь между изменениями мозжечка и аутизмом у детей.
Дети с гидроцефалией в раннем возрасте беспокойны, плохо спят, характерен монотонный крик, усиление рефлексов, плавающие движения глаз и их закатывание, выраженность сосудов роговицы, заметная подкожная венозная сеть по мере роста размеров головки. Спонтанная двигательная активность новорожденных может быть ослаблена, возможны судороги и тетрапарез из-за гипертонуса мышц.
В более старшем возрасте становится заметным отставание в психическом и интеллектуальном развитии, дети не могут обучаться, быстро устают, плохо усваивают новую информацию, что делает процесс адаптации крайне затруднительным. В тяжелых случаях обучение невозможно совсем, в связи с чем ребенок нуждается в постоянной посторонней помощи, уходе и рассмотрении вопроса об инвалидности.
Моторное развитие заметно замедлено. При тяжелых формах аномалии дети не могут своевременно научиться переворачиваться, ползать, садиться и ходить, не удерживают взгляд на игрушках, быстро устают и часто плачут. Возможны нарушения питания с гипотрофией, общее снижение иммунитета, частые инфекционные заболевания.
Сочетание порока нервной системы с другими аномалиями развития органов предрасполагает к серьезным осложнениям, в числе которых не только мозговая дисфункция, слабоумие, судорожный синдром, но и сердечная недостаточность, склонность к пневмониям при пороках сердца, хроническая почечная недостаточность и уремия при врожденном поликистозе, что усугубляет явления отека мозга и может послужить причиной гибели пациента.
При тяжелой окклюзионной гидроцефалии смерть может наступить в раннем младенчестве от отека головного мозга, фатальных аритмий, остановки дыхания на фоне компрессии стволовых структур, тяжелой пневмонии и других инфекционных осложнений.
У взрослых возможно постепенное нарастание гидроцефалии с краниалгиями, снижением памяти и внимания, раздражительностью, склонностью к депрессии, утренней тошнотой и рвотой на высоте головной боли. В тяжелых случаях бывает судорожный синдром. Возможны проблемы с координацией и выполнением мелких движений, неуверенность при ходьбе, зрительные расстройства.
Диагностика и лечение
Диагностика синдрома Денди-Уокера основывается на результатах ультразвукового осмотра, при этом важно обнаружить аномалию еще во время эмбрионального развития. УЗИ становится информативным после 18 недели гестации, но в некоторых случаях заподозрить патологию можно и раньше — уже на 14-15 неделях эмбрионального развития.

Диагностическими критериями при аномалии задней черепной ямки считаются:
- Наличие крупной кистозной полости, включающей четвертый мозговой желудочек, в задней части черепа;
- Отсутствие или аномальное развитие червя мозжечка;
- Гипоплазия мозжечковых гемисфер, наличие широкой щели между ними;
- Расширение желудочковой системы (гидроцефалия).
Для постановки диагноза синдрома Денди-Уокера необходимы:
- УЗИ (нейросонография);
- МРТ для определения анатомических особенностей четвертого желудочка мозга;
- Консультация офтальмолога;
- Осмотр нейрохирурга;
- УЗИ сердца для исключения врожденных аномалий;
- Консультация генетика и определение кариотипа при возможных генетических мутациях.
Лечение патологии определяется симптоматикой и тяжестью проявлений. Если гидроцефалии нет, а внутричерепное давление в пределах нормы, то оправдано динамическое наблюдение невролога, педиатра или нейрохирурга, каких-либо медикаментов не требуется.

шунтирование для нивелирования гидроцефалии
При нарастании гидроцефалии и внутричерепного давления показаны шунтирующие хирургические операции для отвода ликвора из черепа в грудную или брюшную полость. Медикаментозное лечение включает применение диуретиков (диакарб, маннитол), ноотропных средств (пирацетам, пантогам), антиконвульсантов (депакин).
В случае гипертонуса показаны физиотерапевтические и водные процедуры, массаж, специальные упражнения. Важен тщательный уход и постоянное наблюдение за малышом, создание спокойной обстановки при беспокойном поведении и нарушениях сна.
При тяжелых формах течения патологии с отставанием в интеллектуальном развитии детям показана работа с дефектологами-педагогами, психологом по индивидуальной программе, исключающей избыток информации и умственное перенапряжение.
Прогноз при синдроме Денди-Уокера зависит от ряда причин: времени установления диагноза, наличия других пороков и хромосомных болезней, степени окклюзии ликворных путей. Смертность и заболеваемость после рождения выше в тех случаях, когда аномалия сочетается с другими дефектами и обнаружена до рождения малыша.
Гидроцефалия и внутричерепная гипертензия — ключевые моменты в определении прогноза, которые влияют и на развитие пациента, и на продолжительность и качество его жизни. В случае изолированного поражения мозга без признаков гидроцефалии прогноз благоприятный. Ребенок может развиваться по возрасту, а иногда аномалия и вовсе выявляется у взрослых при обследовании по поводу других причин.
В связи с тем, что причины порока так и не выяснены, проводить специфическую профилактику не представляется возможным. Конечно, нужно соблюдать здоровый образ жизни, особенно, женщинам, планирующим беременность или уже забеременевшим с исключением вредных привычек, неблагоприятных влияний внешней среды. Важно своевременно выявить и пролечить цитомегаловирусную инфекцию, герпес, а в случае краснухи, которой женщина заболела при беременности, врачи предложат аборт по медицинским показаниям из-за высокого риска сочетанных пороков.
Решать вопрос о сохранении беременности в том случае, если синдром возник случайно, у плода абсолютно здоровой женщины, придется будущей маме и ее семье. Решение всегда дается сложно, но следует знать, что аномалия нервной системы и нормальное развитие и рост ребенка — скорее, исключение из правил.
В абсолютном большинстве случаев детям и родителям приходится бороться с гидроцефалией, зачастую требуется не одна дорогостоящая и сложная операция, тогда как ее эффективность и прогноз все равно могут оставаться сомнительными.
Видео: примеры детей с синдромом Денди-Уокера
НАСЛЕДСТВЕННАЯ ГИДРОЦЕФАЛИЯ (СИНДРОМ ДЕНДИ-УОКЕРА) КЛИНИЧЕСКИЙ СЛУЧАЙ
Лысова Ю.В.
Научный руководитель: к.м.н., доцент Нечаев В.Н.
Резюме
Количество врожденных пороков развития в последнее десятилетие заметно увеличилось. Пороки развития нервной системы суммарно занимают третье место в структуре аномалий развития после врожденной патологии сердечно-сосудистой и мочевыводящей систем, причем около 80% этих заболеваний представлены гидроцефалией различного генеза [1]. Среди большого количества возможных аномалий одним из наиболее тяжелых по своим последствиям считается синдром Денди Уокера.
Ключевые слова
Статья
Данный синдром был впервые описан американским нейрохирургом Уолтером Денди в 1921 году и Эрлом Уокером в 1944 г. Среди живорождённых детей частота встречаемости от 1:5000 до 1:25000, а среди детей с врождённой гидроцефалией колеблется от 3,5 до 12% [2].
Синдром Денди-Уокера (Dandy-Walker Syndrome) – это порок развития головного мозга (мозжечка и окружающих его ликворных пространств), для которого характерна триада симптомов: гипотрофия червя мозжечка и/или полушарий мозжечка, кисты задней черепной ямки, гидроцефалия различной степени. Данный синдром по МКБ Х кодируется в рубрике аномалий развития под шифром Q 03.1, как атрезия отверстий Мажанди и Люшка [3, 4].
Согласно современным представлениям этиология синдрома Денди-Уокера чрезвычайно гетерогенна, так как в его возникновении принимают участие разные факторы: наследственные (хромосомные и генные) и экзогенные тератогены; предполагается, что определенную роль играют и такие факторы как вирусная инфекция (ЦМВ, краснуха); приём алкоголя, диабет беременно. В 1/3 – 1/2 случаев синдром Денди-Уокера сочетается с другими различными врожденными синдромами (табл. 1) [2].
В 70% случаев порок сочетается и с другими аномалиями головного мозга — агенезией мозолистого тела, энцефалоцеле, полимикрогирией, агирией, гетеротопией серого вещества, а также с поражениями других органов и систем (полидактилией, синдактилией, врожденными пороками сердца, поликистозом почек, расщелинами неба и др.) [5].
Среди основных гипотез возникновения синдрома Денди-Уокера можно выделить следующие:
– остановка эмбрионального развития в процессе формирования ромбовидного мозга;
– атрезия выходного отверстия из IV желудочка при отсроченном открытии отверстия Мажанди;
– возникновение сосудистого сплетения IV желудочка в середине тонкой крыши ромбовидного мозга.
Специалисты пренатальной функциональной диагностики выделяют полную и неполную, а также закрытую и открытую формы синдрома Денди-Уокера. Полная форма характеризуется агенезией червя мозжечка и наличием явной коммуникации между IV желудочком и кистой в области большой цистерны. Неполная форма – это частичная агенезия нижней части червя мозжечка, в связи с чем, коммуникация IV желудочка с кистой большой цистерны прослеживается не на всем протяжении червя. Открытая и закрытая формы различаются наличием или отсутствием окклюзии отверстий Люшка и Мажанди и сообщением желудочка с подпаутинным пространством (рис. 1, 2) [6].
Синдром Денди-Уокера характеризуется выраженным клиническим полиморфизмом: от практически нормального постнатального развития до тяжелой инвалидности и даже гибели ребенка. Перинатальные исходы во многом зависят от глубины поражения ЦНС (прогрессирующая гидроцефалия), а также от наличия сочетанной патологии, которая в 60 – 75% случаев сопровождает данный синдром [2, 5].
Прогноз для жизни и здоровья при синдроме Денди — Уокера зависит от наличия сочетанных аномалий развития, хромосомных аномалий и срока диагностики. По данным литературы, показатели постнатальной заболеваемости и смертности выше в тех случаях, когда синдром диагностирован в пренатальном периоде, а не постнатально [2] .
Лечение данного заболевания симптоматическое. При наличии признаков нарастающей внутричерепной гипертензии проводят шунтирующие операции. Исход часто летальный, в 90% случаев это первые годы жизни [7].
Под наблюдением находился доношенный новорождённый мальчик Л., родившийся от 3 беременности, протекавшей на фоне поздней постановки на учёт (в 30 недель). С 35 недель гестации отмечалось многоводие и впервые по данным ультразвукового исследования выявлена аномалия развития плода – мальформация Денди-Уокера.
Картина пренатального УЗИ в 35 недель гестации: форма головы аномальная (клубникообразная). Кости при надавливании датчиком не деформируются. Расширение боковых желудочков, передние рога до 10 мм справа и слева. Полость прозрачной перегородки до 9,9 мм. Задние рога – 11 мм, слева – 12 мм. Отмечено расширение IVжелудочка до 12 мм. Имеется гипоплазия червя мозжечка, расширение большой цистерны до 12 мм.
Ребенок от третьих срочных родов в головном предлежании. Околоплодные воды светло-жёлтые в объеме двух литров. Масса тела ребёнка при рождении составила 2890 г, рост – 49 см., окружность головы – 35 см., груди – 33 см. Оценка по шкале Апгар 4 – 7 баллов.
Состояние ребёнка после рождения тяжелое, из родильного зала поступил в ОРИТН, за счёт дыхательной недостаточности III степени и выраженной неврологической симптоматики. C момента поступления находился на ИВЛ, с третьих суток жизни был переведен на респираторную поддержку в режиме СРАР, а с 7 суток жизни на спонтанное дыхание. Оксигенотерапия проводилась по показаниям. Энтеральное питание начато с первых суток в трофическом объеме с последующим расширением, усваивал.
Неврологически: положение ребёнка вынужденное на боку с запрокинутой головой, крик монотонный, имеются плавающие движения глазных яблок, симптом «заходящего солнца», непостоянный горизонтальный нистагм. Неправильная форма черепа, увеличенная в размерах мозговая часть, нарастание окружности головы в динамике за 2 недели составило 2,5 см. Отмечалось расхождение костей черепа, швы открыты до 0,5 см, с увеличением размеров большого родничка до 5 × 8 см. Зрачки средней величины, фотореакция сохранена. Спонтанная двигательная активность и мышечный тонус снижены. Дистония в конечностях. Физиологические рефлексы угнетены, сухожильные снижены, симметричные.
Обращало на себя внимание наличие двусторонней косолапости с варусной деформацией обеих стоп, кистей и фаланг пальцев, гипертелоризма, седловидной переносицы, нависающего лба и затылка, широкого языка, короткой шеи (рис. 3).
На 14 сутки жизни, после стабилизации состояния, ребёнок был переведён на второй этап лечения, в отделение патологии новорожденных для дальнейшего наблюдения и лечения.
Картина нейросонографии в первые сутки жизни: в задней черепной ямке при коронарных и сагиттальном сканированиях наблюдается крупное анэхогенное («кистовидное») образование, включающее расширенные III и IV желудочек; полушария мозжечка резко уменьшены, червь не определяется; мозжечковый намет смещен вверх.
В динамике по данным НСГ выявлена перивентрикулярная ишемия, подтверждён синдром Денди-Уокера, вентрикуломегалия (как часть симптомокомплекса) и повышенная резистентность сосудов мозга.
Ребёнок был осмотрен неврологом, окулистом, генетиком, выставлен диагноз: Врожденный порок развития ЦНС – синдром Денди Уокера. Гипоксически-ишемическое поражение ЦНС. После осмотра нейрохирурга выставлен диагноз: ВПР ЦНС синдром Денди-Уокера, гидроцефальный синдром.
На момент осмотра в нейрохирургическом лечении не нуждался, были даны рекомендации по уходу и лечению.
По результатам ДЭХО-КГ выявлен врожденный порок сердца: комбинированный стеноз легочной артерии (клапанно-подклапанный), ДМПП со сбросом слева направо. Открытое овальное окно диаметром 2,0 мм.
Ребенок был консультирован кардиологом и кардиохирургом, даны рекомендации.
По данным УЗИ брюшной полости и почек патологии не выявлено.
После осмотра врача-ортопеда была подтверждена врожденная двусторонняя косолапость.
В связи со стабилизацией состояния ребёнка, после проведённого обследования и лечения, а также отказа матери от родительских прав, мальчик на 57 сутки жизни был переведён в дом ребёнка города Маркса.
В заключение следует сказать о том, что проведённое настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку встречается ни так часто в повседневной практике врача.
Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое клиническое наблюдение, представляет определенные сложности. По моему мнению, в подобных ситуациях оправдана постановка синдромального диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования. Точный нозологический диагноз важен не только для генетического анализа, медико-генетического консультирования, но и, прежде всего, для профилактики и лечения. Без достоверного клинического диагноза невозможен анализ факторов и их теоретическое осмысление.
Литература
1. Кириллова Е.А., Никифорова О.К., Жученко Н.А. и др. Мониторинг врожденных пороков развития у новорожденных // Российский вестник перинатологии и педиатрии. — 2000. — № 1. — С. 18-21.
2. Барашнев Ю.И. Перинатальная неврология. – М.: Триада-Х, 2001.- С. 181-232.
3. Бочков Н.П. Наследственные болезни. Национальное руководство. М.: ГЭОТАР-Медиа, 2012.- С. 128-145.
4. Неонатология (национальное руководство) / Под ред. Н.И.Володина. – М., ГЭОТАР-Медиа, 2007; 847 с.
5. Наследственные болезни / Под ред. Л.О.Бадаляна. – Т.: Медицина, 2002.- С. 138.
6. Пренатальная диагностика наследственных и врожденных болезней / Под ред. Э.К.Айламазяна, В.С.Баранова. – М.: Триада-Х, 2007. – С. 11-148.
7. Барашнев Ю.И., Бахарев В.А., Новиков П.В. Диагностика и лечение врожденных и наследственных заболеваний у детей. М.: Триада-Х, 2004.- С.12-87.
Таблицы
Синдромы, с которыми сочетается гидроцефалия Денди-Уокера
Синдромы
Клинические признаки
Этиология
Различные хромосомные аномалии
Поражение различных органов и систем
Хромосомные делеции, дупликации, трисомии, триплоидии
Лиссэнцефалия, ретинальная дисплазия, аномалия глаз, энцефалоцеле, миопатии и др.
Мутации генов POMT1 (локализации 9g34.1), POMT2 (14g24.3), FKTN (9g31) и др. Тип наследования аутосомно-рецессивный
3С синдром (краниомозжечково-сердечная дисплазия, Ritscher-Schinzel синдром)
Задержка роста, пороки сердца (септальные дефекты), гипертелоризм и др.
Ген не установлен. Тип наследования аутосомно-рецессивный
Артериальные аномалии, включая коарктацию аорты, дефекты сердца, глаз (микроофтальмия).
Синдром Денди-Уокера: клинические проявления и лечение
Синдром Денди – Уокера — врожденная аномалия развития, характеризующаяся нарушениями строения мозжечка и путей оттока ликвора. Первые признаки болезни выявляются при проведении планового ультразвукового исследования в период беременности. У детей после рождения нарушена моторика, присутствуют явления гидроцефалии, а также другие симптомы. Для устранения признаков расстройства назначаются медикаменты, снижающие тонус мышц и уменьшающие внутричерепное давление. Возможно проведение хирургических операций для улучшения оттока спинномозговой жидкости.
Причины развития
Выявить причину развития заболевания не всегда удается. Специалисты отмечают ряд признаков, приводящих к симптомам болезни:
- увеличение объема четвертого желудочка, что приводит к скоплению большого объема ликвора;
- нарушения оттока спинномозговой жидкости в результате зарастания или полного отсутствия путей для этого;
- объемные опухолевые образования в задней черепной ямке;
- нарушение строения мозжечка. У детей выявляется увеличение объема полушарий на фоне атрофии червя.
Указанные изменения в строении структур головного мозга связаны с генетическими аномалиями у плода. Они возникают на фоне перенесенных внутриутробных инфекций (краснуха, цитомегаловирусная патология, герпес), аутоиммунных или обменных нарушений у беременной. При длительной интоксикации (табакокурение, алкоголизм) в период беременности риск возникновения синдрома Денди – Уокера также увеличивается.
Клинические проявления
Первые признаки болезни могут быть выявлены на УЗИ во время прохождения беременной женщиной ультразвукового скрининга. При этом специалист определяет следующие патологические изменения:
- уменьшение размера мозжечка, в первую очередь, червя, расположенного между его полушариями;
- кистозное образование в задней черепной ямке;
- увеличение размера четвертого желудочка.
Эти признаки могут наблюдаться при других наследственных заболеваниях — синдроме Марфана, Дауна и пр. Дифференциальная диагностика в период внутриутробного развития ограничена.
После рождения симптоматика синдрома Денди – Уокера представлена следующим образом:
- диспропорции головы ребенка. Она имеет увеличенный размер, что связано с гидроцефалией;
- краниотабес, характеризующийся размягчением костей в области родничков;
- увеличение размеров родничков;
- гидроцефалия, приводящая к утренним головным болям, тошноте, рвоте и слабому крику ребенка;
- нарушения формирования челюсти и носовых раковин;
- нистагм, проявляющийся непроизвольными колебательными движениями глазных яблок;
- нарушения согласованных движений рук и ног, а также мелкой моторики;
- общее беспокойство.
Дети в возрасте более 1 года характеризуются тревожностью и раздражительностью. После пробуждения они жалуются на головную боль. У них появляются тошнота, рвота и моторные нарушения. В процессе взросления формируются умственная отсталость и задержка психомоторного развития. Патология часто сопровождается аномалиями развития внутренних органов: сердца, легких и др.
Негативные последствия
Осложнения заболевания развиваются на фоне основных симптомов патологии. К негативным последствиям болезни относятся:
- задержка психомоторного развития разной степени выраженности;
- умственная отсталость;
- очаговая неврологическая симптоматика: гипертонус мышц рук и ног, гиперкинез отдельных мышечных групп, нарушенная координация движений;
- хроническая сердечная недостаточность;
- заболевания мочевыделительной системы и др.
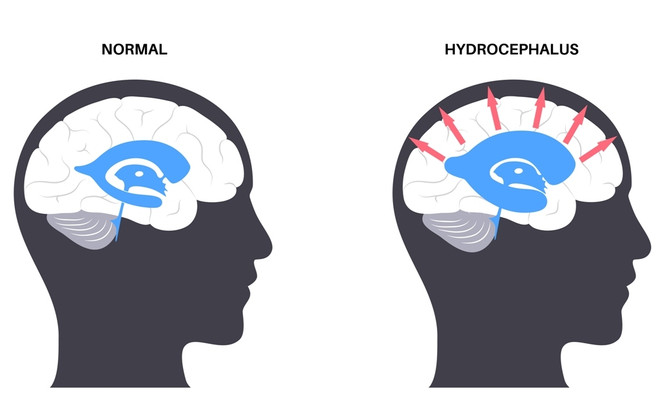
Синдром Денди – Уокера приводит к инвалидности с рождения. 40–50% пациентов не доживает до 6 месяцев. Аналогичный прогноз наблюдается при других наследственных заболеваниях — синдроме Ретта и пр.
Диагностические мероприятия
Основная задача врача — проведение пренатальной диагностики патологии. Ультразвуковое исследование плода позволяет заподозрить болезнь и провести дифференциальную диагностику с другими врожденными аномалиями (синдром Эдвардса и др.). Подтвержденный диагноз является показанием для прерывания беременности при согласии женщины. После рождения ребенка диагностические мероприятия проводятся по определенному алгоритму:
- Сбор имеющихся жалоб и внешний осмотр ребенка. Специалист выявляет аномалии строения костей черепа, увеличение размера родничков и головы, а также неврологическую симптоматику. Последняя представлена гипертонусом мышц и нарушениями мелкой моторики.
- При беседе с родителями, выявляются факторы риска развития патологии — перенесенные в период беременности инфекционные заболевания, алкоголизм, наркомания и др.
- Проводится УЗИ структур центральной нервной системы и внутренних органов. Обследование позволяет выявить аномалии развития и определить тактику их лечения.
- Компьютерная или магнитно-резонансная томография используются для исследования состояния внутренних органов. Методы заменяют УЗИ, так как обладают большей диагностической ценностью для врачей.
- Консультации с педиатром, неврологом, кардиологом, офтальмологом и другими специалистами при наличии показаний.
Интерпретировать результаты проведенных исследований должен только врач. Неправильная постановка диагноза — причина прогрессирования нарушений работы головного мозга и внутренних органов, приводящих к тяжелым осложнениям.
Подходы к лечению
Полное выздоровление невозможно. Терапия позволяет увеличить продолжительность жизни и предупредить развитие осложнений заболевания. Она включает в себя консервативные методы и хирургические вмешательства.
Операции направлены на восстановление оттока ликвора. С этой целью врачи проводят шунтирование — создают дополнительные пути для спинномозговой жидкости. Вмешательство на четвертом желудочке головного мозга также уменьшает признаки гидроцефалии. Кроме этого, операции проводятся при пороках сердца, почек и других внутренних органов.
Лечение медикаментами устраняет основные проявления болезни. Общий прогноз не меняется. При выраженном мышечном гипертонусе назначают препараты для его уменьшения. Кроме того, врач может выписать другие медикаменты:
- кортикостероидные гормоны — уменьшают количество образующейся спинномозговой жидкости;
- препараты, расширяющие кровеносные сосуды, снижают внутричерепное давление за счет улучшения оттока ликвора;
- диуретические средства;
- бета-блокаторы уменьшают отечность нервной ткани и внутричерепное давление.
К средствам поддерживающей терапии относят:
- обезболивающие препараты из группы нестероидных средств, позволяющих снизить выраженность боли на фоне внутричерепной гипертензии;
- аминокислотные средства, улучшающие обменные процессы в нервных клетках и защищающие их от повреждений;
- успокоительные препараты, используемые при повышенной возбудимости и гиперактивности ребенка.
Если у больного наблюдаются сопутствующие поражения внутренних органов, то врач подбирает средства для уменьшения их симптоматики.
Важно помнить, что все препараты имеют определенные показания и противопоказания, которые необходимо учитывать при их назначении. Препараты назначает только врач!
В терапии используют немедикаментозные подходы: лечебный массаж и физкультуру, а также водные процедуры. Указанные методы нормализуют мышечный тонус, снимают спазм мускулатуры и уменьшают выраженность болевого синдрома.
Как предупредить заболевание?
Специфическая профилактика наследственных болезней отсутствует. Врачи рекомендуют беременным женщинам соблюдать следующие принципы в период вынашивания плода:
- избегать воздействие токсических веществ на работе, а также отказаться от курения, употребления спиртных напитков и наркомании;
- своевременно обращаться в женскую консультацию для наблюдения у гинеколога;
- соблюдать профилактику инфекционных заболеваний. Если женщина имеет хроническую цитомегаловирусную или герпетическую инфекцию, в период беременности следует поддерживать высокую активность собственного иммунитета;
- не пропускать ультразвуковой скрининг и другие назначения врача.
Простые советы помогают снизить риск развития наследственных патологий, в том числе синдрома Клайнфельтера и др. При наличии в семье генетических болезней в период планирования беременности рекомендуется проконсультироваться с врачом-генетиком. Он проведет обследование женщины и мужчины, составив список рекомендаций, снижающих вероятность рождения ребенка с аномалиями развития.
Специфическая профилактика отсутствует. Основная задача родителей — пройти подготовку к беременности и устранить факторы риска.
Синдром Денди – Уокера — тяжелое наследственное заболевание с неблагоприятным прогнозом. Около 50% новорожденных не доживает до полугода и умирает на фоне недостаточности внутренних органов или тяжелого поражения головного мозга.
Своевременное внутриутробное выявление болезни с помощью УЗИ-скриннига позволяет провести медицинское прерывание беременности и предупредить рождение больного ребенка. Эффективная терапия болезни невозможна и носит симптоматический, поддерживающий характер.

